
Table of Contents
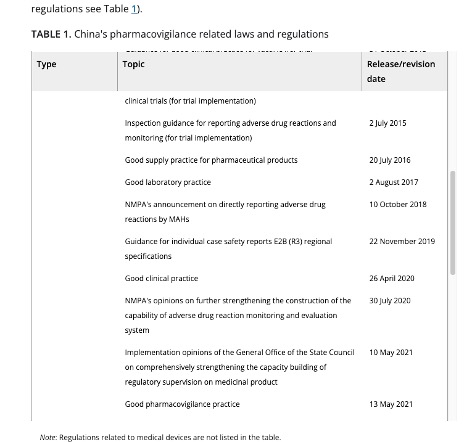
China’s Good Pharmacovigilance Practices (GVP)
An essential Guide to China’s Good Pharmacovigilance Practices (GVP)
China’s National Medical Products Administration (NMPA), formerly known
asCFDA, released the China Good Pharmacovigilance Practice (GVP)
guidelines in May 2021. These China GVP guidelines are designed to improve
and align China’s pharmacovigilance regulations and practices with
international standards set by the International Council for Harmonization
(ICH).
The China GVP guidelines consist of nine chapters and 134 articles,
providing a comprehensive framework for marketing authorization holders
(MAHs) and drug registration applicants in China to establish and
implement a robust pharmacovigilance (PV) system. These guidelines cover
important aspects such as reporting adverse drug reactions (ADRs), signal
detection, PV master files, and strengthening of passive monitoring.
By adhering to the China GVP guidelines, MAHs and clinical drug trial
sponsors can ensure compliance with China’s PV regulations, enhance drug
safety for patients, and minimize risks associated with adverse drug
reactions. Implementing a comprehensive PV system in accordance with
global standards not only promotes patient safety but also contributes to
the overall quality and credibility of the pharmaceutical industry in
China.
The China Good Pharmacovigilance Practices (GVP) guidelines contain
the following table of content:
Chapter 1 – Overview
Chapter 2 – Quality Management
- Section 1 Basic Requirement
- Section 2 Internal audit
- Section 3 Entrusting Management
Chapter 3 – Institutional Personnel and Resources
- Section 1 Organisation/Institutions
- Section 2 Personnel training
- Section 3 Facility and Resources
Chapter 4 – Monitoring and Reporting
- Section 1 Information Collection
- Section 2 Report Assessment and Processing
- Section 3 Report Submission
Chapter 5 – Risk Identification and Assessment
- Section 1 Signal Detection
- Section 2 Risk Evaluation
- Section 3 A Post-Authorisation Safety Study (PASS)
- Section 4 Periodic Safety Update Report
Chapter 6 – Risk Control
- Section 1 Risk Control Measures
- Section 2 Risk Communication
- Section 3 Pharmacovigilance Plan
Chapter 7 – Management of Documents, Records and Data
- Section 1 Policy and Procedural Documents
- Section 2 Pharmacovigilance System Master File (PSMF)
- Section 3 Record and Data
Chapter 8 – Pharmacovigilance during Clinical Trial
- Section 1 Basic Requirement
- Section 2 Risk Monitoring, Identification, Evaluation and Control
Chapter 9 – Supplementary Provisions
To download the full copy of China GVP guidelines, you may click
here.</a >
China Pharmacovigilance History
Pharmacovigilance in China: Milestones and Advancements
China pharmacovigilance regulatory reform has a history dating back to
1980’s with the building of ADR monitoring networks in China to more
significant evolution after China joined ICH in 2017 which is now the
foundation and commitment towards an internationally harmonised PV system.
China strengthened pharmacovigilance policies with the introduction of
Drug Administration Law in 2019 which set out the framework for developing
China Good Pharmacovigilance Practices (GVP) in 2021.
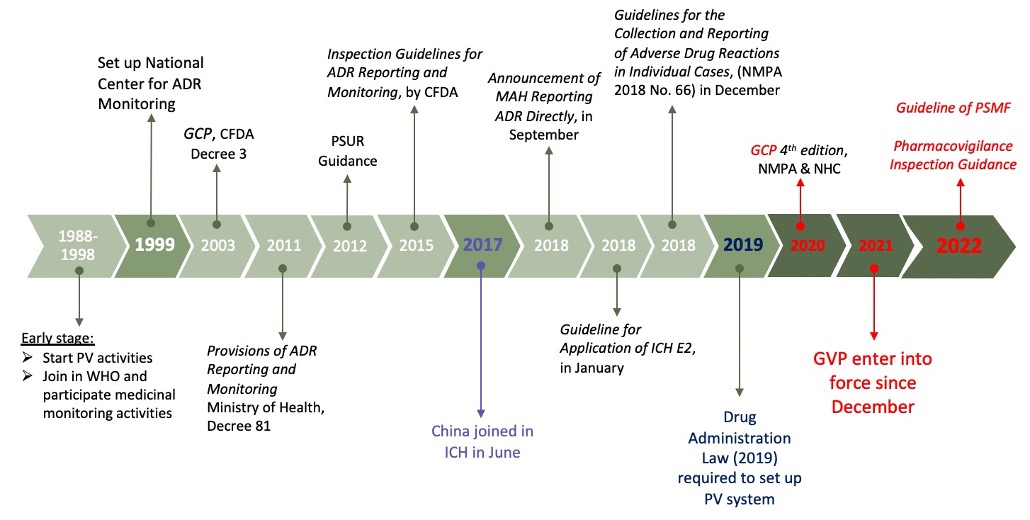
- 1950 to 1998: Building of ADR monitoring network in China.
- 1988: Ministry of Health initiates a pilot project on ADR monitoring
involving 10 hospitals in China. - 1994: 66 hospitals in 26 provinces in China designated as key hospitals
for ADR monitoring, marking a significant step towards a comprehensive
system. - Pre-2017: Regulations and implementation of pharmacovigilance in China
not mature nor systematic. - September 2018: China NMPA releases Announcement No. 66 mandating MAHs
to report ADRs through NADRMS based on the principle of reporting on
suspicion in CHina. - 2019: China Drug Administration Law released, requiring pharmaceutical
companies to establish pharmacovigilance and annual reporting systems in
China. - 2020: China GCP revised with the release of the fourth edition by NMPA
and NHC. - May 2021: China GVP 2021 is officially released and comes into force on
December 1st, marking a new chapter for PV in China. - 2022: Official China guidance document on writing pharmacovigilance
systems is published, further standardizing the framework and reducing
the difficulty of building PV systems.
China PV Policy Trend
In the near to medium term, the Chinese authorities is expected to
continue to accelerate and enforce GVP and pharmacovifilance regulations
to improve drug safety and patient outcomes.
CIOMS and PV
China Pharmacovigilance and CIOMS: Building a Strong Framework for Drug
Safety

Revised for SEO:
CIOMS plays a crucial role in the field of pharmacovigilance by
coordinating and publishing guidelines on drug safety, working with
representatives from the global biomedical scientific community. The
organization’s programs focus on key areas of biomedicine, including
bioethics, health policy, ethics, human values, drug development and use,
and international nomenclature of diseases. Since the 1980s, CIOMS has
been running projects that address drug safety, and its working groups
have published several guidelines for practice. These guidelines are
influential in shaping pharmacovigilance practice worldwide and are widely
used, although they have no legal or regulatory mandate. CIOMS also
developed the CIOMS Form I, a standard format for reporting suspected
adverse reactions to medical products that has proved valuable in practice
and is still widely used.
The organisation was founded by the WHO and UNESCO, the United Nations
Educational, Scientific and Cultural Organisation, in 1949. It is an
independent not-for-profit body which is not affiliated to, or funded by,
any single government or nation. The organisation brings together
representatives from the “biomedical scientific community” worldwide,
aiming to encourage and facilitate international biomedical scientific
activities whilst maintaining a relationship with the United Nations
organisation (particularly WHO and UNESCO)
The CIOMS working groups have published a number of practice guidelines,
including
- I – Internatonal reporting form (also known as CIOMS I Form)
- II – Periodic Safety Update Reports (PSUR)
- III – Guideline for Preparing Core Clinical Safety Information (CCSI)
on Drugs - IV – Benefit-risk assessments for marketed drugs
- VI – Management of Safety Information from Clinical Trials
- VII – Development safety update reports (DSUR)
- VIII -Signal Detection(Publication: Practical Aspects of Signal
Detection in Pharmacovigilance)
In addition, CIOMS was involved in publishing an initiative to standardise
the use of medical terms associated with adverse drug reactions. However,
this has not been widely accepted in pharmacovigilance practice.
Reference:
</a >https://cioms.ch/publications/?filterby=pharmacovigilance
ICH and PV
Understanding the Role of ICH in Drug Safety and Pharmacovigilance

Revised for SEO:
The International Council for Harmonisation (ICH) was established in 1990
as a collaboration between regulatory authorities and industry
professionals to ensure the safety, quality, and efficacy of medicines
intended for human use. The need for greater uniformity in testing and
safety regulations across regions led to the formation of ICH, which has
been able to develop practice guidelines that meet global standards
through working groups comprised of representatives from regulatory
authorities, pharmaceutical industries, and organizations such as the
World Health Organization.
The ICH has established numerous documents outlining safety standards for
both pre-clinical and clinical stages of drug development. Pre-clinical
guidelines are identified with an “S” designation, while clinical safety
guidelines are marked as “E” for “Efficacy.” Each clinical safety
guideline has a unique identifying code, which is periodically revised to
reflect updated standards. The ICH has developed several clinical safety
guidelines that have reached “step 4” status and provide detailed
expectations for reporting clinical data to regulatory authorities.
E2A Clinical Safety Data Management provides definitions and standards for
the expedited reporting of adverse drug reactions (ADRs) to regulatory
authorities to ensure patient safety. E2B (R2) Maintenance of the Clinical
Safety Data Management including Data Elements for Transmission of
Individual Case Safety Reports provides a standardized format for the
maintenance and transmission of individual case safety reports (ICSRs) to
regulatory authorities to ensure timely and accurate reporting of ADRs.
E2B (R3) Clinical Safety Data Management: Data Elements for Transmission
of Individual Case Safety Reports updates and expands on the previous E2B
(R2) guideline, providing a more comprehensive list of data elements to be
included in ICSRs.
E2C (R1) Clinical Safety Data Management: Periodic Safety Update Reports
for Marketed Drugs provides guidance on the content, format, and frequency
of periodic safety update reports (PSURs) for marketed drugs, with the aim
of ensuring ongoing assessment of drug safety.
E2C (R2) Periodic Benefit-Risk Evaluation Report provides guidance on the
preparation and submission of periodic benefit-risk evaluation reports
(PBRERs) for marketed drugs, with the aim of ensuring ongoing assessment
of the balance between drug efficacy and safety.
E2D Post-Approval Safety Data Management: Definitions and Standards for
Expedited Reporting provides definitions and standards for the expedited
reporting of ADRs that are identified post-approval, with the aim of
ensuring ongoing monitoring and assessment of drug safety.
E2E Pharmacovigilance Planning provides guidance on the development of
pharmacovigilance plans for new drugs, with the aim of ensuring proactive
monitoring and assessment of drug safety.
E2F Development Safety Update Report provides guidance on the preparation
and submission of development safety update reports (DSURs) for drugs in
clinical development, with the aim of ensuring ongoing assessment of drug
safety during the clinical trial process.
Please note that this page cannot provide detail on the full scope of the
ICH guidelines and the interested reader is referred instead to the source
material which can be found online at the ICH website, detailed in the
references below. Please note that this page should not be considered as
professional pharmacovigilance advice
Application of ICH in China Pharmacovigilance
According to CFDA Announcement on the Application of Secondary Guidelines
of International Conference on Harmonization Technical Requirement for
Registration of Pharmaceuticals for Human Use (No.10, 2018)
- From 1 May 2018, E2A, M1, E2B(R3) applied for SUSAR in Clinical
studies of medicinal products - From 1 July 2018, E2D applied for the reporting of post-marketing ADR
- From 1 July 2019, the requirement of M1 and E2B (R3) can be applied
for post-marketing ADR and from 1 July 2022, above technical
guidelines (M1 and E2BB (R3)) applied to the reporting of
post-marketing ADR.
According to Announcement by NMPA on the Application of 15 ICH Guidelines,
including “E1: Exposure Levels in Populations: Assessing the Clinical
Safety of Drugs for the Long-Term Treatment of Non-Life-Threatening
Diseases” and others (No. [2019] 88)
- E2F will be applicable from the Announcement date (12 Nov 2019)
- E2E: will be applicable to NDA accepted 3 months after the
Announcement date & NDA approved 6 months after the Announcement date
China’s Phamacovigilance Regulators: A Closer Look at NMPA, CDE, and CDR
Exploring the responsibilities of NMPA, CDE, and CDR in China’s PV system

SAMR – State Administration for Market Regulation
The SAMR is a full ministry agency reporting directly to the State Council
of the People’s Republic of China. Under the SAMR is the NMPA, which
regulates clinical trials and pharmaceutical market industry.

NMPA – National Medical Products Administration
China NMPA (Formerly CFDA) is the regulatory authority responsible for
China’s drug surpervision and management, which includes implementing
regulations and guidelines for ensuring drug safety in clinical trials,
marketing authoritisations and pharmacovigilance policies in China.
Other responsibilities:
- Management and supervision of drugs, cosmetics, and medical devices.
- Establishing standards for drugs, cosmetics, and medical devices.
- Managing registration of drugs, cosmetics, and medical devices.
- Handling quality management for all regulated products.
- Risk management of regulated products after listing.
- Managing qualification and registration of licensed pharmacists.
- Inspection and supervision of drugs, cosmetics, and medical devices.
- Handling foreign exchanges and other interactions with international
parties. - Guiding drug supervision and administration departments under
government jurisdiction.

China CDE (Centre for Drug Evaluation)
NMPA’s Center for Drug Evaluation (CDE) is responsible for the evaluation
of drug clinical trial applications, drug marketing authorization
applications, supplementary applications, and overseas drug production
registration applications. China’s Centre for Drug Evaluation (CDE) is
also responsible for pharmacovigilance reporting pre-marketing Adverse
Events, including Serious Adverse Events and Unintended Serious Adverse
Events in China.

CDR-Centre for Drug Reevaluation, NMPA | National Center for ADR
Monitoring</strong >
China’s Centre for Drug Reevaluation (CDR) is responsible for the
regulation of China’s pharmacovigilance, post-marketing pharmacovigilance,
and colleagues are responsible for issuing some technical standards and
guidelines.
ADR Reporting in China: Official System explained

National Adverse Drug Reactions Monitoring System (NADRMS)</strong >
NADR Monitoring System is used for Adverse Events Reporting and
Management:
- Medicinal
- Medical Device
- Cosemetics
- Drug Abuse
Reference:
https://www.adrs.org.cn/</a >
China Direct ADR Reporting System for MAH, NCADRM
Reference:
https://daers.adrs.org.cn/#/login?_k=mi0b3p</a >
National Medical Device Adverse Event Monitoring Information System,
NCADRM</strong >
Reference:
https://maers.adrs.org.cn/console/login.ftl</a >
Cosmetic Adverse Reaction Monitoring System
Reference:https://caers.adrs.org.cn/adrcos/</a >
Drug Abuse reporting and Management Portal
Reference:
http://111.202.232.186/sso/login?service=http%3A%2F%2F111.202.232.186%2FPF%2FcasAuthUser</a >
Inserting regulations like the following format (scrolling)?
Clinical Pharmacovigilance (PV) in China
CDE – Regulatory Authority Responsible for China Clinical
Pharmacovigilance Activities
The Clinical Trials Management Office, which is subordinated to China
Center for Drug Evaluation (CDE), is specifically responsible for the
reception, analysis and evaluation of suspected and unexpected serious
adverse reactions (SUSAR) during clinical trials conducted in China, as
well as development safety update reports (DSURs) during the research and
development of relevant drugs. Following the guidelines of ICH E2A, E2B
and E2F, among others, China CDE has published a series of technical
standards and normative documents based on the actual situation in China
which includes an electronic reporting system for SUSAR cases during
clinical trials.
Pharmacovigilance in clinical trials is the process of monitoring and
assessing the safety of investigational drugs or therapies during the
clinical trial. This is essential to ensure that the benefits of the
treatment outweigh any potential risks or adverse effects. The principle
goal of China pharmacovigilance systems in clinical trials is to identify
and prevent any adverse reactions or unexpected events related to the
investigational product.
China Clinical Pharmacovigilance activities typically include the
following:
- Design and implementation of a China pharmacovigilance plan: This plan
outlines the strategies for collecting and reporting adverse events
(AEs) and serious adverse events (SAEs) during the clinical trial. - China pharmacovigilance adverse event reporting and management: All
AEs and SAEs that occur during the clinical trial must be reported and
managed according to regulatory requirements. - Safety monitoring: Regular safety monitoring is conducted to identify
any potential safety issues that may arise during the clinical trial. - Risk management: Strategies are implemented to mitigate the risks
associated with the investigational product and to ensure the safety
of the trial participants. - Safety reporting in China: All safety data and analyses are compiled
and reported to regulatory authorities, ethics committees, and other
relevant stakeholders.
China pharmacovigilance in clinical trials plays a crucial role in
ensuring the safety of study participants and in providing reliable safety
data for regulatory approval of new drugs or therapies. It also helps to
build trust between the pharmaceutical industry, regulatory agencies, and
the general public.
Navigating SUSAR Reporting in China
China CDE began to receive SUSAR reports from drug registration sponsors
on May 1, 2018. In China SUSAR reporting stands for “Suspected Unexpected
Serious Adverse Reaction” reporting. It refers to the requirement for
sponsors of clinical trials to report any serious adverse reactions that
occur during the trial to the relevant regulatory authorities.
In China serious adverse reaction is defined as any untoward medical
occurrence that results in death, is life-threatening, requires
hospitalization or prolongation of existing hospitalization, results in
persistent or significant disability or incapacity, or is a congenital
anomaly/birth defect.
If such an adverse reaction is considered to be unexpected (i.e. not
consistent with the product’s known safety profile), the sponsor is
required to report it to the China regulatory authorities as a SUSAR. The
purpose of SUSAR reporting is to ensure that the safety of trial
participants is monitored and that any potential risks associated with the
investigational product are identified and communicated to the relevant
authorities.
China SUSAR Reporting
The specific guideline for China SUSAR reporting is “The Standards And Procedures for Expedited Reporting of Safety Data
during Clinical Trials</strong >” which serves as the official guidline document published on 27 Apr
2018, Effective date 1st May 2018.
The applicant must report any unexpected and serious adverse reactions
that are assessed as certain or suspected to be related to the
investigational drug. If the applicant and investigator cannot come to an
agreement on the causality assessment between the adverse event and the
drug, and neither party can rule out a relationship to the investigational
drug, then a expediated reporting should proceed.
The following scenarios generally do not need to be reported as expediated
reporting:
- non-serious adverse events,
- serious adverse events that are not related to the investigational
drug - serious adverse reactions that are expected, and
- when the primary efficacy endpoint is a serious adverse event, the
applicant is not advised to report it as a case safety report (ICSR)
to the national drug regulatory agency.
The applicant is responsible for determining whether to report serious
adverse reactions related to a positive control drug to other drug
manufacturers and/or directly to the China drug regulatory agency. The
applicant must report such events to the drug manufacturer or directly to
the regulatory agency. Adverse events related to a placebo generally do
not meet the criteria for an adverse reaction and do not need to be
reported.
Adaptation to ICH-E2B(R3)
The content of case safety reports for unexpected serious adverse
reactions should follow the requirements outlined in the ICH “E2B(R3):
Management of Clinical Safety Data – Data Elements for Transmission of
Individual Case Safety Reports” and use the ICH “M1: MedDRA” to code
relevant terms.
The applicant is responsible for monitoring the safety information of
clinical trials and reporting serious adverse events. The applicant must
appoint a dedicated person to be responsible for clinical trial safety
information monitoring and serious adverse event report management,
establish standard operating procedures for clinical trial safety
information monitoring and serious adverse event reporting, and train all
relevant personnel. The applicant must also be aware of the latest safety
information during the clinical trial process, conduct timely safety risk
assessments, inform relevant trial stakeholders of relevant information,
and be responsible for reporting unexpected serious adverse reactions in a
timely manner.
China SUSAR Reporting Deadlines
After learning of a serious adverse event, the applicant should
immediately conduct a comprehensive analysis, evaluation, and judgment of
the event. According to the nature (category) of the serious adverse
event, the applicant should report it to the Center for Drug Evaluation
(CDE), NMPA quickly within the following time limits:
- For unexpected serious adverse reactions that cause death or endanger
life, the applicant should report as soon as possible after learning of
it, but no later than 7 days, and report and improve follow-up
information within the following 8 days.
Note: The day the applicant first learns of the event is Day 0.
- For unexpected serious adverse reactions that do not cause death or
endanger life, the applicant should report as soon as possible after
learning of it, but no later than 15 days.
The start time for quick reporting is the approval date of the clinical
trial/national drug regulatory authority’s implicit permission start date,
and the end time is the date of the last follow-up of the last subject in
the country. Serious adverse events that occur between the end of the
clinical trial or follow-up and obtaining the evaluation and approval
conclusion should be reported by the investigator to the applicant, and if
they are unexpected serious adverse reactions, quick reporting should also
be conducted.
After the first report, the applicant should continue to track serious
adverse reactions and report new information or changes to the previous
report in a timely manner in the form of follow-up reports. The reporting
time limit is 15 days from obtaining new information.
Fatal or life-threatening SUSAR for China
In China for fatal or life-threatening SUSAR, the applicant should report
as soon as possible but no later than 7 days after the first report is
made and submit a follow-up report with as much information as possible
within 8 days of the first report. For subsequent reporting of new
information in the form of a follow-up report or changes to the previous
report, the time limit for reporting is 15 days from the date the new
information was obtained.
The Center for Drug Evaluation (CDE), NMPA receives the individual case
safety reports submitted by the applicant during the drug clinical trial
period in an e-submission format that complies with ICH E2B (R3), and
conducts analysis and evaluation. When necessary, opinions on modifying
the trial plan, suspending or terminating the drug clinical trial, etc.
will be put forward according to relevant standards.
In addition to individual safety reports of unexpected serious adverse
reactions, the applicant should also report other potential serious safety
risk information to the national drug regulatory authority as soon as
possible, and make medical and scientific judgments on each situation.
Generally speaking, information that obviously affects the drug
risk-benefit assessment or may consider changing the drug use, or affects
the overall drug development process, belongs to this category, such as:
- The occurrence rate of known serious adverse reactions has increased,
and it is judged to be clinically important; - There is a significant harm to the exposed population, such as the
drug being ineffective in treating life-threatening diseases; - Significant safety findings (such as carcinogenicity) in recently
completed animal experiments.
The applicant should also report unexpected serious adverse reactions
related to the trial drug and other potential serious safety risk
information obtained from other sources to the Chinese drug regulatory
authority quickly.
SUSAR Report in Chinese Language
Both domestic and foreign individual safety reports and reports of other
potential serious safety risks should be reported in Chinese.
The acceptance number of the drug application clinical trial should be
clearly marked in both individual safety reports and reports of other
potential serious safety risks.
Reference:
1.The Standards And Procedures for Expedited Reporting of Safety Data
during Clinical Trials Announcement of the publication of the Standards
and Procedures for Expedited Reporting of Safety Data during Drug Clinical
Trials
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
China DSUR Reporting
In China, DSUR also stands for Development Safety Update Report. China CDE
opened the DSUR reporting submission channel on its official website to
receive DSUR reports from drug registration sponsors on April 26, 2019,.
In 2020, overall 1775 copies of DSUR reports were submitted to CDE by the
sponsors.
DSUR in China is a required regulatory document that provides a
comprehensive summary of the safety profile of a drug under investigation
during a clinical trial. The DSUR is submitted at regular intervals to the
CDE and is intended to be a tool for ensuring that the safety of trial
subjects is being appropriately monitored and that any emerging safety
concerns are being addressed.
The China DSUR includes a description of any adverse events or safety
issues that have been identified during the reporting period, along with
an assessment of their severity and potential impact on the study or the
overall safety profile of the drug. It also includes information on any
changes that have been made to the trial protocol or study design, as well
as any actions taken to address safety concerns or manage risks to study
participants.
The DSUR is typically required for all clinical trials of investigational
drugs, and may be requested by regulatory authorities at any stage of the
clinical trial process. The contents are reviewed by the Chinese
authorities as part of their ongoing assessment of the safety and efficacy
of the drug, and may be used to inform decisions about whether to allow
the clinical trial to continue or whether to require additional safety
measures.
RMP (Risk Identification, Evaluation & Control) in China
RMP outlines the potential risks associated with a medicinal product and
the measures that are in place to minimize or manage those risks. The
purpose of an RMP is to identify, characterize, and assess the known and
potential risks of a medicinal product, and to determine whether
additional measures are necessary to mitigate these risks.
The Origin and Implementation of Pharmacovigilance Planning in China
As early as 12 November, 2019, the NMPA published a notice on 15 ICH
guidelines (No. 88 of 2019), requiring that new drug marketing
applications (NDAs) accepted three months after the date of publication,
and new drug marketing applications approved six months after the date of
publication starting to implement the ICH E2E: Pharmacovigilance Planning.
The ICH E2E Guidelines focus on the submission of a Safety Specification
and Pharmacovigilance Planning by the Regulatory Authority at the time of
application for marketing authorization.
Safety Specification
The safety specification for China should be “a synopsis of important
identified risks, important potential risks, and important missing
information about the drug” and should begin with a summary of
epidemiological information about the target indication.
Regardless of the indication and target population, a risk should be
classified as important if it has the following characteristics:
- The risk leads to serious consequences when it occurs. such as death,
disability or a serious impact on the quality of life of the person
using the drug. - requires a high proportion of clinical 1000 precautions.
- presents a significant challenge to current clinical practice.
Significant risks may not affect all drug-using populations but are
only highly prevalent in drug-users with certain characteristics.
Applicants are advised to assess the risk causation scale,
preventability and its impact on the benefit-risk balance and as an
important reference for the development of risk control measures.
On July 1, 2020, the CDE published the “M4 Module 1 Administrative
Document and Drug Information”, which explicitly requires applicants to
submit NDA information “1.8.3 Risk Management Plan (RMP)”, including
Pharmacovigilance Plan and Risk minimization Measures.
In the ICH E2E Guidelines and relevant Chinese laws, regulations, and
guidelines, the terms “Pharmacovigilance Plan”, “Risk Management Plan”,
and “Risk Analysis and Management Plan” are considered as one concept due
to the refinement of laws and translation.
Pharmacovigilance Plan
Pharmacovigilance plans should include both:
- Routine pharmacovigilance activities
- Additional pharmacovigilance activities
If not specifically requested by the CDE, it is recommended that the first
post-marketing evaluation of the pharmacovigilance programme/RMP be
conducted approximately 2 years after the product is launched and may
include, but is not limited to:
- The implementation of the pharmacovigilance plan and RMP.
- Whether the cumulative post-marketing data obtained influenced the
judgement of product risk - Whether the pharmacovigilance activities undertaken are adequate or no
longer applicable - The evaluation of the effectiveness of risk minimisation measures;
- Whether the pharmacovigilance plan and RMP affects the accessibility
of the product or places an unnecessary burden on the healthcare
system.
China Pharmacovigilance Plan China referencing ICH E2E as follows:
Routine Pharmacovigilance Practices
Routine pharmacovigilance should be conducted for all medicinal products,
regardless of whether or not additional actions are appropriate as part of
a Pharmacovigilance Plan. This routine pharmacovigilance should include
the following:
- Systems and processes that ensure that information about all suspected
adverse reactions that are reported to the personnel of the company
are collected and collated in an accessible manner; - The preparation of reports for Chinese regulatory authorities:
Expedited adverse drug reaction (ADR) reports
Periodic Safety Update Reports (PSURs) - Continuous monitoring of the safety profile of approved products
including signal detection, issue evaluation, updating of labeling,
and liaison with regulatory authorities; - Other requirements, as defined by local regulations.
Action Plan for Safety Issues
The Plan for each important safety issue should be presented and justified
according to the following structure:
- Safety issue;
- Objective of proposed action(s);
- Action(s) proposed;
- Rationale for proposed action(s);
- Monitoring by the sponsor for safety issue and proposed action(s);
- Milestones for evaluation and reporting.
Summary of Actions to be Completed, Including Milestones
An overall Pharmacovigilance Plan for the product bringing together the
actions for all individual safety issues should be presented. Whereas
section 3.1.3 suggests presenting an action plan by ongoing safety issue,
for this section the Pharmacovigilance Plan for the product should be
organised in terms of the actions to be undertaken and their milestones.
The reason for this is that one proposed action (e.g., a prospective
safety cohort study) could address more than one of the identified issues.
It is recommended that milestones for completion of studies and other
evaluations, and for submission of safety results, be included in the
Pharmacovigilance Plan. In developing these milestones one should
consider when:
- Exposure to the product will have reached a level sufficient to
allow potential identification/characterisation of the AEs/ADRs of
concern or resolution of a particular concern; and/or - The results of ongoing or proposed safety studies are expected to be
available.
These milestones might be aligned with regulatory milestones (e.g.,
PSURs, annual reassessment and license renewals) and used to revise the
Pharmacovigilance Plan.
Post-marketing PV in China
Definition
Post-marketing pharmacovigilance (PV) refers to the collection,
monitoring, assessment, and reporting of adverse drug reactions (ADRs)
that occur after a drug has been approved and marketed for use in the
general population. Post-marketing PV activities are carried out by
regulatory authorities, pharmaceutical companies, and healthcare
professionals to ensure that drugs remain safe for use in the general
population. The ultimate goal of post-marketing PV is to identify and
minimize the risks associated with the use of medicinal products, thereby
ensuring the safety and well-being of patients.
ADR Monitoring Network in China and Main Responsible Competent
Authority</strong >
China’s drug adverse reaction monitoring network is divided into four
levels, including the National Centre for Drug Re-evaluation (CDR), 34
provincial drug adverse reaction monitoring centres, and hundreds of
municipal and county-level institutions responsible for adverse drug
reaction monitoring.1 These institutions monitor and evaluate medical
device-related adverse events, cosmetic adverse reactions, and drug abuse,
in addition to adverse drug reactions. The role of China’s adverse drug
reaction monitoring agencies has gradually been shifted to
pharmacovigilance management.
At present, CADRMS incorporates a direct reporting system for ADRs for
MAHs, a reporting system for ADRs for medical institutions and
enterprises, a monitoring system for medical device-related adverse
events, a monitoring system for adverse cosmetic reactions and a reporting
system for drug abuse.2 In addition, CADRMS has functional modules for
data analyses, signal detection, potential alert management and periodic
safety update report management.3 In recent years, CDR has explored and
constructed an active monitoring system for drug safety based on
electronic medical records from medical institutions.
In 2010, the Ministry of Health and the National Medical Products
Administration formulated the National Guideline for the Surveillance of
Suspected Adverse Events following Immunization, which clearly outlines
the reporting requirements for Adverse Events Following Immunization
(AEFI) for post-approval vaccines. According to this guideline,
responsible reporting units and reporters are required to report suspected
AEFI to their local county-level Centre for Disease Control and Prevention
agency where the vaccine recipients reside.4 China has gradually
established and enhanced its AEFI monitoring information management
system, and the AEFI case reporting network was completed on a national
level in 2008.5 The Chinese Centres for Disease Control and Prevention
established the AEFI Monitoring System, which shares information with the
Drug Adverse Reaction Monitoring Agency.
Reference:
1. Zhao Y, Wang TS, Li GY, Sun S. Pharmacovigilance in China: Develop-
ment and challenges. Int J Clin Pharmacol. 2018;40(4):823-831.
2. National Center for ADR Monitoring of China. The Chinese Adverse Drug
Reaction Monitoring System. Available at http://www.adrs.org.cn/.
3. Song HB, Shen C. Thoughts on the use of electronic health data in the
safety research of marketed drugs. China Food Drug Administrat Mag.
2020;36-47.
4. National Surveillance Program for Suspected Abnormal Response of
Vaccination. Chin J Vacc Immun. 2011;17:72-81.
5. Liu DW, Wu WD, Li KL, et al. Surveillance of adverse events following
immunization in China: Past, present, and future. Vaccine. 2015;
33(32):4041-4046.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Individual Adverse Drug Reactions Reporting and Monitoring in China
What is an Individual Case Safety Report (ICSR) in China?
So, what is an ICSR? According to the ICH E2 series of related guideline
documents ICSRs are complete information provided by the reporter at a
point in time to describe an event or incident of concern. The report may
include information relating to a subject or a group of subjects.
According to Chinese Guidelines for the Collection and Reporting of
Individual Adverse Drug Reactions1 A valid report is defined to be
consisted of following four elements:
- Identifiable patient
- Identifiable reporter
- Suspected drug
- Adverse reaction.
If all four elements are incomplete, the report is considered invalid and
should be added before reporting. “Identifiable” means that the presence
of the patient and the reporter can be confirmed. A patient is considered
identifiable when one or more of the following are available: name or
initials, sex, age (or age group, e.g., adolescent, adult, elderly), date
of birth, other identifying code for the patient. The initial reporter who
provided the case information or the relevant person contacted to obtain
the case information should be identifiable. For case reports from the
Internet, the identifiability of the reporter depends on the ability to
verify the existence of the patient and the reporter, e.g., by providing a
valid email address or other contact information.
Reference:
1. Guidelines for the Collection and Reporting of Individual Adverse Drug
Reactions,
Announcement by the NMPA on the Issuance of Guidelines for the Collection
and Reporting of Individual Adverse Drug Reactions (No. [2018] 131)
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Sources & Collection of Adverse Events in China
According to China GVP1, cases are collected from different sources:
Medical Institutions
Approach like visits, email, telephone, and fax to collect information on
AE occurring in clinical settings from medical personnel on a regular
basis, keep detailed records, and establish and maintain an adverse drug
reaction information file. Doctors could also inform the AE information
collected from the patients to the Medical representative who subsequently
inform the MAH.
Drug Distributors
The holder or its distributor should ensure that the pharmaceutical
retailer is aware of the effective ways to report AE to it, develop an AE
collection plan, and train the pharmacist or other personnel based in the
pharmacy,
I.e., Pharmacist received AE information feedbacked from the patient that
purchased drug on this pharmacy. The pharmacy then send email to PV public
mail or call the hotline number for AE complaint.
Phone calls & Complaints
Calls from the patient that saw the hotline indicated in either Package
Insert or outer packaging of the drug or on the MAH websites.
Academic Literature
AE information identified through literature filtered in literature
databases (from literature screening process)
Internet Relevant Channels
MAH should regularly visit the websites they sponsor or manage to collect
possible cases of AE. In principle, MAH are not required to search
external websites, but if MAH are informed of AE in external websites,
they should assess whether to report them.
The MAH should use the company website/portal to collect information on
AE, i.e., by creating a dedicated pathway for ADR reporting on the
website, providing guidance on how to report, reporting forms and
reporting content, and publishing complete and up-to-date product
instructions. Graphic media, digital media, social media/platforms
initiated or managed by the MAH are also a source of individual adverse
drug reactions, i.e., collected using corporate social medial (i.e.
Instagram or WeChat public accounts, microblogs, forums, etc.
Post-marketing Research & Projects
Individual AE identified in post-marketing studies (including those
conducted outside of China) or organised data collection projects
initiated by the company should be reported as required, such as clinical
trials, non-interventional epidemiological studies, focused drug
monitoring, patient support projects, market research or other marketing
projects, etc. AE identified in post-marketing studies or projects should,
in principle, be reported by the MAH to the regulatory authority, but the
holder shall not interfere with the reporting behaviour of the study or
project collaborators for any reason or by any means.
Regulatory Authority
If consumer or healthcare professional directly report adverse events to
regulatory authority (i.e., FDA, EMA etc..) not to marketing holder
(pharmaceutical company), then those reports will be shared to marketing
holders by regulatory authorities. This type of cases which were received
by companies from regulatory authority are to be handled as regulatory
authority cases. Feedback cases downloaded from the ADR monitoring system.
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)Should you have any queries or
seeking for consultation, please contact us via online chat or email.
Spontaneous AE Reporting in China
A spontaneous report is a direct communication of adverse event
information by healthcare professionals or consumers to a company,
regulatory authority, or other organization (e.g., WHO, Regional Centres,
Poison Control Centre).
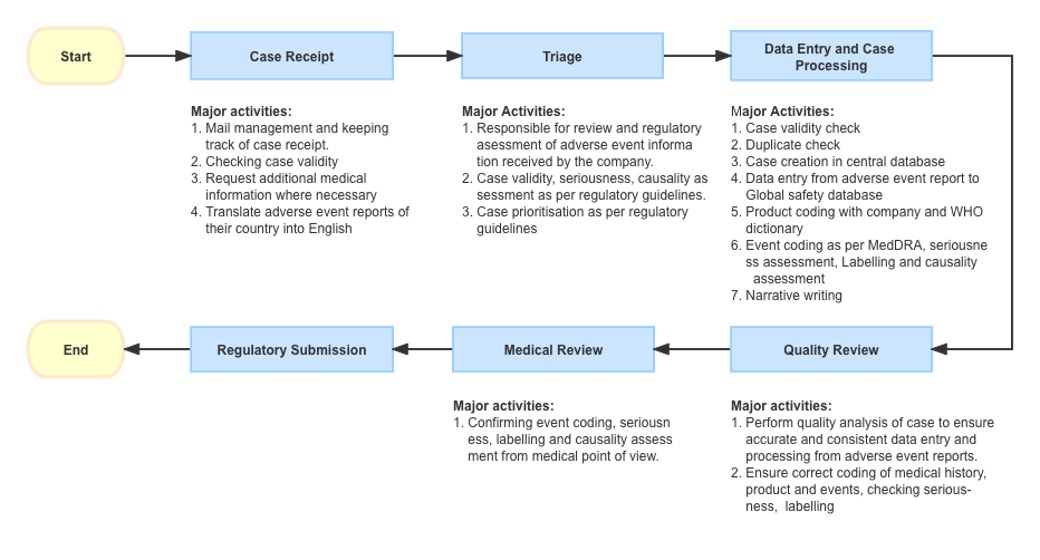
Reporting to Pharmaceutical companies: We can even report events directly
to manufacturer of product by sending emails/fax or by telephone (details
are available in google), some companies have call centres to collect
adverse events.
The Call centre personnel received reporting of an AE/AEs from call or an
email from a patient’s relative. After an initial confirmation of the case
authenticity and validity assessment i.e., the report has identifiable
reporter information, an identifiable patient, suspect product/s and AEs,
the report will be triaged to PV personnel for further processing.
PV team collect the reported events and perform below assessments (using
global sponsor’s database for processing and manual submission to RA):
- Initial seriousness of the event was evaluated
- Assess the reason for event occurrence
- Understanding if it is new event or already known information
- The source document that includes all the communication contents and
reporting information will be translated (if necessary) - Input collected report information in the PV safety database and
attaching the source doc + exercise due diligence schedule to collect
missing if necessary - Fill out the information collected as shown below (ArisG System
database used for example) - General information, patient information, protocol/project information
(for Research and clinical trial products), product information,
adverse event, reporter information - After initial review of the case processing workflow, the report will
be going to next workflow where it is further reviewed and narrative
will be written in medical format, the events are coded using Medical
Dictionary for Regulatory Activities (MedDRA), causality is assessed,
eventually releasing completed CIOMS I form. - Submitting information to regulatory authority based on the translated
CIOMS form - Save the receipt of submission report (pdf) in local file for
inspection/audit purposes. - Update the tracker.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Feedback Case Reporting in China
Log in the ADR monitoring system.
Click the feedback data button and download the feedback data in excel
form
Translate into English to generate an ICSR with all the safety information
(if applicable), Input in the PV Safety database and then following the
process the same as that of Spontaneous case reporting until the
submission to RA completed.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Regulatory Submission
Per the GVP requirement, Spontaneous and feedback case reports can be
submitted through ADR monitoring system either manually or through the
National Safety Database that is completed interface with the ADR
monitoring system.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Medical Review & Narrative Writing
Medical review and narrative writing are crucial components of
pharmacovigilance. Medical review involves analysing the safety data
generated from clinical trials or post-marketing surveillance to identify
potential safety signals and assess the overall benefit-risk profile of a
drug. This process requires a deep understanding of the drug’s mechanism
of action, clinical pharmacology, and the medical condition it is intended
to treat. Medical reviewers must be able to critically evaluate adverse
event reports and determine their causality, seriousness, and
expectedness. They also need to assess the impact of the adverse event on
the patient’s health and well-being and determine if any additional
monitoring or risk minimization measures are necessary.
Narrative writing involves summarizing the relevant clinical and safety
information in a concise and coherent manner to provide a clear picture of
the patient’s medical history and the circumstances surrounding the
adverse event. A well-written narrative should provide a chronological
account of the patient’s medical history, including any relevant
comorbidities, concomitant medications, and previous drug exposure. It
should also describe the onset and progression of the adverse event,
including the time to onset, duration, and severity, as well as any
interventions or outcomes. The narrative should be clear, concise, and
free of ambiguity or unnecessary detail.
Effective medical review and narrative writing are critical for
identifying and managing potential safety risks associated with drugs.
Medical reviewers and writers must be highly trained professionals with a
strong background in pharmacology, clinical medicine, and data analysis.
They play a vital role in ensuring the safety of patients and the
continued success of drug development programs.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Aggregate Reporting in China
Periodic Safety Update Report/Periodic Benefit Risk Evaluation Report –
Requirement in China
Periodic safety update report (PSUR) provides a periodic and comprehensive
assessment of the worldwide safety data of a marketed drug. Over-time it
was recognized that the risk of the marketed drug should be assessed in
the light of its benefits and change in the risk estimate overtime.
Consequently, the report name was changed to Periodic Benefit-Risk
Evaluation Report (PBRER).
According to China GVP1,
Chapter V Risk Identification and Assessment
Section 4 Periodic
Safety Report
Article 79 The periodic safety update report shall be based on the work
carried out by the holders during the reporting period. A comprehensive
and in-depth review, summary and analysis of the collected safety
information shall be conducted, and the format and content shall meet the
requirements of “Guidance for Writing Periodic Safety Update Reports of
Drugs”.
Article 80 For innovative drugs and improved new drugs a periodic safety
update report shall be submitted every 1 year from the date of obtaining
the approval documents until the first re- registration, and then every 5
years. For other types of drugs, the periodic safety update report shall
generally be submitted every 5 years from the date of obtaining the
approval documents. If the drug regulatory authority or the adverse drug
reaction monitoring agency requires otherwise, submission shall be made in
accordance with the requirements
Article 81 The data summarization time of the periodic safety update
report starts from the date when the drug approval document is first
obtained or starts from the first approval date for marketing of the drug
around the world (i.e., the International Birth Day (IBD). Integrity and
continuity should be maintained during the periodic safety update report
data coverage period).
Article 82 Periodic safety update reports shall be submitted through the
National Adverse Drug Reaction Monitoring System after approval by the
pharmacovigilance responsible person.
Article 83 The holders shall deal with and respond to the review opinions
on the periodic safety update reports in a timely manner. For analysis and
assessment requirements for specific safety issues, in addition to
separate submission as required by the drug regulatory authority or the
adverse drug reaction monitoring agency, it should also be analyzed and
evaluated in the next periodic safety update report.
Article 84 The holders may submit a periodic benefit-risk assessment
report instead of a periodic safety update report. The writing format and
submission requirements shall comply with the relevant guidelines of the
International Conference on Harmonization of Technical Requirements for
Registration of Pharmaceutical for Human Use. Other requirements are the
same as those for periodic safety update reports.
Article 85 The risk assessment in the periodic safety update report shall
be based on all uses of the drug.When carrying out a benefit-risk
assessment, the assessment of effectiveness shall include data from
clinical trials and data obtained during actual use for the approved
indication. The comprehensive benefit-risk assessment should be based on
the approved indications, combined with the risks in the actual use of the
drug.
Article 86 Unless otherwise required by the drug regulatory authority,
periodic safety update reports do not need to be submitted for the
following drugs or products managed as drugs: raw materials, in vitro
diagnostic reagents, Traditional Chinese medicinal materials, and
Traditional Chinese medicine decoction pieces.According to NMPA
Announcement on the application of the ICH – E2C(R2): PBRER2, MAH can
submit PBRER following the ICH guidelines.
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
2. NMPA Announcement on the application of the ICH Guidelines, E2C(R2):
PBRER (No. [2020]86)
Should you have any queries or seeking for consultation, please contact
us via online chat or email.</strong >
Official FAQ related to PSUR in China
Which products are not required to submit a PSUR?
For APIs, excipients and in-vitro diagnostic reagents that are subject to
the approval number, PSURs do not need to be submitted.
For Chinese
herbal medicines, Chinese herbal drinks and imported Chinese herbal
medicines that are subject to approval number control, it is not required
to submit PSUR. For products that are manufactured by domestic
pharmaceutical manufacturers entrusted from abroad (e.g. certified by
EU/FDA and other relevant countries/regions, in compliance with the laws
and regulations of the commissioning country) but have not obtained the
approval documents from China, it is not required to submit PSUR.
How should an overseas pharmaceutical manufacturer of an imported drug,
which does not have a drug manufacturer in China but has an office or an
agency appointed by it in China and a general agent for its products in
China, submit a PSUR?
The overseas pharmaceutical manufacturer of the drug should designate its
office (or agent) or general agent in China as the safety representative
of the drug in China, to fulfil the responsibility of reporting adverse
drug reactions and submitting PSURs for the drug. After obtaining the
authorisation from the overseas pharmaceutical manufacturer, the
representative office (or agency) or general agent in the country will
register with the Adverse Drug Reaction Monitoring Platform through the
provincial centre where the safety representative is located and then
submit the PSUR.
Can a marketing authorisation holder use a Periodic Benefit-Risk
Evaluation Report (PBRER) to be submitted instead of a PSUR? What are
the specific requirements if a PBRER is submitted?</strong >
Yes. If a PBRER is submitted, the format and timeline for submission apply
to the ICH E2C (R2): Periodic Benefit-Risk Evaluation Report (PBRER), i.e.
PBRERs with a reporting period of 1 year or less should be submitted
within 70 days of the Data Lock Point (DLP) and within 90 days for those
with a reporting period of 1 year or more. The guidelines and their
questions and answers are available in the ICH section of the Centre for
Drug Evaluation website.
Other questions can be found in the Guidelines for Writing Periodic Safety
Update Reports (PBRERs) and Frequently Asked Questions and Answers (Q&A).
The PBRER for imported medicines should be translated into Chinese and
submitted together with the original English PBRER, except for the annexes
to the PBRER.
Fore more questions please leave your contact emails and we will provide
you the Translated version.
Reference:
1. PSUR Frequent Question Answered (Q&A) answered 1 – 5
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Signal Management in China: An Overview of Regulatory Requirements
Overview of Signal Management
Signal Management is a set of activities performed to determine whether,
based on an examination of individual case safety reports (ICSRs),
aggregated data from active surveillance systems or studies, scientific
literature information or other data sources, there are new risks
associated with an active substance or a medicinal product or whether
known risks have changed, as well as any related recommendations,
decisions, communications, and tracking.The Signal Management consists of
- Signal Detection
- Signal Validation
- Signal Prioritization
- Signal Assessment
Regulatory Guidance of Signal Management
According to China GVP1, the scope and requirement for signal management
is listed below:
Chapter V Risk Identification and Assessment
Section 1 Signal Detection
Article 55 The holders should carry out signal detection on the suspected
adverse drug reaction information collected through various channels to
discover new drug safety risks in a timely manner.
Article 56 The holders should select an appropriate, scientific, and
effective signal detection method according to its own situation and
product characteristics. The signal detection methods can be manual
detection methods such as individual adverse drug reaction report review,
case series evaluation, and case report summary analysis, or
computer-aided detection methods such as data mining.
Article 57 The frequency of signal detection shall be reasonably
determined according to relevant factors such as the time of drug
marketing, drug characteristics, and risk characteristics. For newly
marketed innovative drugs, improved new drugs and other varieties that
attention required by the drug regulatory authorities or drug adverse drug
reaction monitoring authority, at or above the provincial level, the
frequency of signal detection shall be increased.
Article 58 When carrying out signal detection, the holders should pay
special attention to the following signals:
- Adverse drug reactions not mentioned in the drug package insert,
especially serious adverse drug reactions. - Adverse drug reactions mentioned in the drug package insert, but the
frequency of occurrence、 severity,etc. have increased significantly. - Suspected new adverse drug reactions caused by drug-drug, drug-device,
and drug-food interactions. - Suspected new medications in special populations or changes of
medications in known special populations. - Suspected adverse reactions show clustering characteristics, and the
correlation with drug quality cannot be ruled out.
Article 59 The holders shall determine the priority of signals, and
prioritize the evaluation of signals that may affect the benefit-risk
balance of the product or have an impact on public health. The following
factors can be considered for signal priority determination:
- The seriousness, severity, outcome, reversibility, and preventability
of adverse drug reactions; - Patient exposure status and expected frequency of adverse drug
reactions; - Patient exposure in high-risk populations and populations with
different medication modes; - The impact of interruption of treatment on patients and the
availability of other treatment options; - Anticipated risk control measures that may be taken;
- Signals applicable to other same class drugs.
Article 60 The holders shall comprehensively summarize relevant
information, evaluate the detected signals, and comprehensively determine
whether the signals constitute a new drug safety risk.
Relevant
information includes individual adverse drug reaction reports ((including
feedback reports from adverse drug reaction monitoring agency), clinical
study data, literature reports, epidemiological information on adverse
drug reactions or diseases, non-clinical study information, medical
database information, relevant information released by drug regulatory
authority or adverse drug reaction monitoring agency, etc. If necessary,
the holders can obtain more information by conducting drug post-marketing
safety studies, etc.
Article 61 If the holders aware of or discovers that multiple suspected
adverse reactions with similar clinical manifestations in the same batch
of the same drug (or adjacent batches) in a short period of time,
presenting clustering characteristics, a case analysis and investigation
shall be carried out in a timely manner.
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Responsible Person for PharmacoVigilance (RPPV)
The Qualified Person for Pharmacovigilance (QPPV) is a key role in the
field of drug safety and regulatory compliance. The QPPV is responsible
for overseeing the pharmacovigilance system of a pharmaceutical company or
a marketing authorization holder, ensuring that all adverse events and
safety issues related to their products are effectively monitored,
evaluated, reported and managed in compliance with relevant regulations.
The QPPV acts as the main point of contact between the company and
regulatory authorities, providing oversight and guidance to their team of
pharmacovigilance professionals. They are responsible for ensuring the
safety of the company’s products throughout their lifecycle, from clinical
trials to post-marketing surveillance.
The QPPV must have a thorough understanding of the company’s products and
the regulations governing their use, as well as the ability to effectively
communicate safety information to both internal and external stakeholders.
They must also maintain a continuous monitoring of safety data and
emerging safety concerns, and ensure that appropriate actions are taken in
a timely manner to protect patient safety.
In China the similar role is named Responsible Person for
Pharmacovigilance (RPPV). The main scope, responsibility, and regulatory
requirement of RPPV is illustrated in China GVP.
See also Comparison of EU-QPPV and China RPPV Section for more detailed
China RPPV requirement.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Pharmacovigilance Audit and Inspections in China
Internal Audit
Pharmacovigilance audit is a vital process that reviews the drug safety
system to ensure it meets regulations, guidelines, and standard operating
procedures. It involves internal or external auditors who are independent
of the system. Internal auditors, part of the safety or quality assurance
team, conduct periodic reviews, while external auditors provide an
objective assessment.
The audit evaluates various aspects, including the organizational
structure, standard procedures, safety database, signal detection, risk
management, and adverse event reporting. It assesses data quality,
identifies system gaps, and provides recommendations for improvement.
Compliance with regulations and guidelines is a key focus of
pharmacovigilance audits. The audit team ensures adherence to Good
Pharmacovigilance Practice (GVP) and International Council for
Harmonization (ICH) guidelines, ultimately enhancing drug safety. China
GVP also described the audit requirement1:
Chapter II Quality Management
Section 2 Internal Audits
Article 11 Holders shall conduct periodic internal audits (hereinafter
referred to as “internal audits”) to review various systems, procedures,
and their implementation status, and to assess the suitability, adequacy,
and effectiveness of the pharmacovigilance system. When any significant
changes occur to the pharmacovigilance system, internal audits should be
carried out in a timely manner. The internal audit can be carried out
independently, systematically, and comprehensively by the holder’s
designated personnel, or by external personnel or experts.
Article 12 An audit plan shall be developed before the internal audit. The
plan should include internal audit objectives, scope, methods, standards,
auditors, audit records and reporting requirements, etc… While
developing the plan, key pharmacovigilance activities, key positions, and
previous audit results, etc. should be considered.
Article 13 Records shall be kept for internal audits, including the basic
information, content and results of the audit, and a written report shall
be formed.
Article 14 For the findings identified in the internal audit, holders
shall investigate the root cause and take corresponding corrective and
preventive actions. The corrective and preventive actions should be
followed-up and assessed.
Reference: 1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Regulatory Inspections
The overarching authority responsible for drug and pharmacovigilance
inspections is China’s National Medical Products Administrations (NMPA).
The pharmacovigilance audit and inspection work are conducted in groups
formed by the Drug Inspection Institutions. The complete inspection is
fulfilled with the support of the following four institutions:
- Drug Testing Institutions
- Centre for Drug Evaluation (CDE)
- Centre for Drug Reevaluation (CDR)
- National Centre for ADR Monitoring.
In addition, the legal departments will be involved upon legal issues.
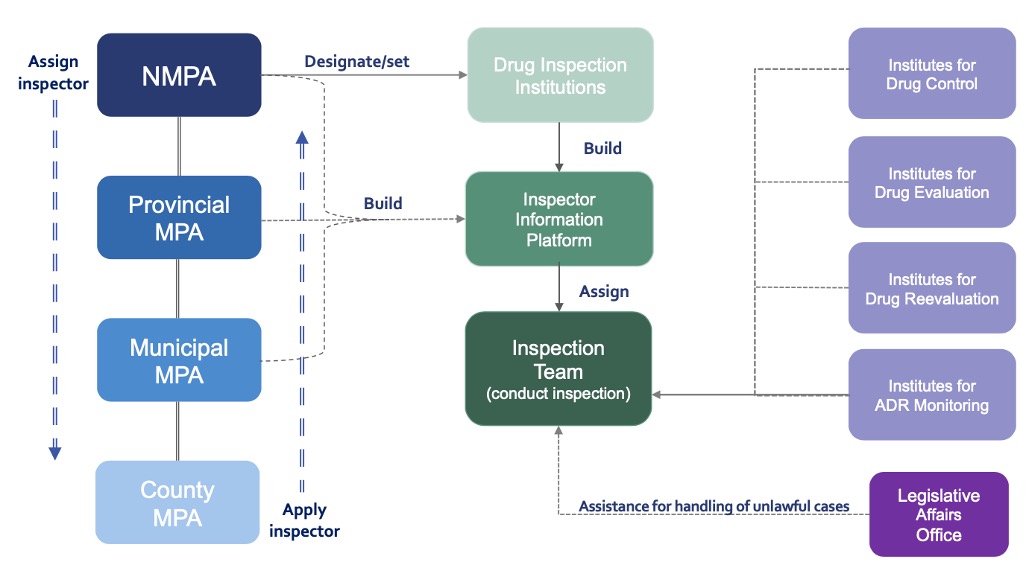
Authority for Supervision of Drugs
- NMPA and Local MPA including Provincial, Municipal, and County level are
responsible to set up and designate pharmaceutical and pharmacovigilance
inspection institutions in accordance with law. - NMPA shall be responsible for forming annual supervision and inspection
plan, arranging inspection tasks, or organizing inspections, and work
according to the Report on Comprehensive Evaluation of Drug Inspection
and relevant evidence materials. - NMPA shall set up a professional team of specialized drug inspectors,
implement multi-level & classification system for inspectors, formulate
the standards of duties and duties of inspectors at different levels as
well as requirements for comprehensive capability, and establish strict
criteria for relevant positions. - NMPA and the drug inspection institution is responsible for establishing
inspector database and inspector information platform to realize the
information sharing and coordination of inspectors at all levels from
national to county-level.
Institutions for Drug Inspection
- Drug inspection institutions carry out inspections according to China’s
laws and regulations on drug supervision before issuing the
Comprehensive Evaluation Report on Drug Inspection and be responsible
for daily management of professional inspector team and implementation
of inspection plans and tasks. - Other departments such as drug inspection, review, evaluation, and
adverse drug reaction (ADR) monitoring established shall provide
technical support during the inspection. - Drug inspection institutions shall establish a quality management system
to continuously improve the quality of drug inspection work. - The drug supervision authority or drug inspection institution is
responsible for establishing inspector database and inspector
information platform to promote the information sharing and coordination
of inspection within national, provincial, and county levels.
Drug Inspection Group
- The inspection group should be set up by the pharmacovigilance
inspection team to carry out the audit. - Usually, an inspection group consists of more than 2 qualified
inspectors - The team leader is responsible for the inspection team. When necessary,
experts in relevant fields can be selected to participate in the
inspection
Authority Inspection Key Points
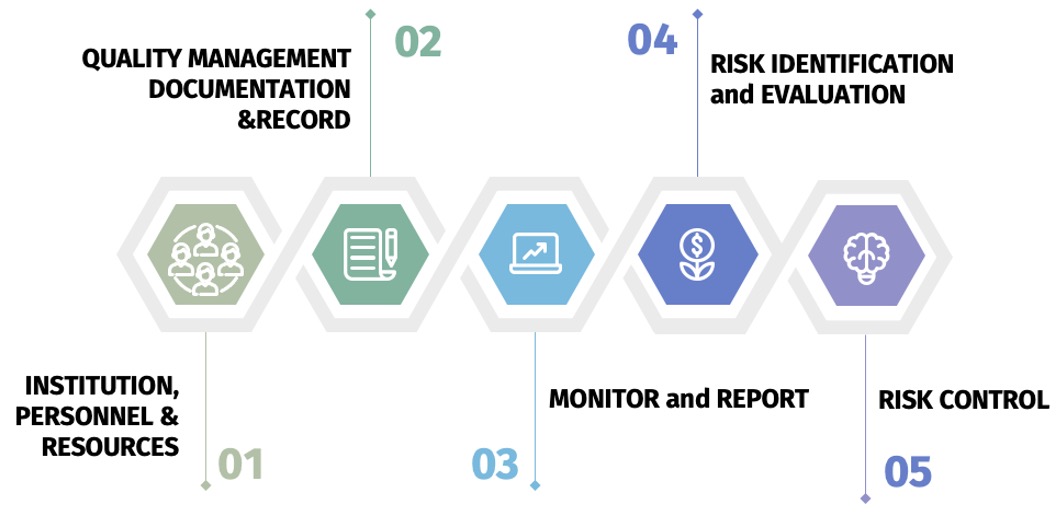
According to Guidelines for Pharmacovigilance Inspections1 released by the
NMPA, a total of 100 checking items are listed. 12 of which are critical
and 40 are major points.
This is the 5 key points for authority inspection.
- Institution, personnel and resources, refers to 22 articles from GVP
- Quality management, documentation and record, refers to 28 articles
- Monitor and report, refers to 17 articles.
- Risk identification and evaluation, refers to 19 articles.
- Risk control, refers to 14 articles.
Throughout the first year of 2022, the authorities overseeing drug
testing, review, evaluation, and adverse drug reaction (ADR) monitoring
focused their attention primarily on the first two points. These areas
were given priority due to their direct accessibility and evaluative
nature. In addition, the authorities made efforts to understand the
operational practices within pharmaceutical companies. As we enter 2023,
inspection groups have gained valuable experience, which will be reflected
in their increased emphasis on activities such as signal detection,
reporting, and risk control, among others.
The following is part of Pharmacovigilance Inspection Check Points in the
official Guidelines for Pharmacovigilance Inspections.
For full PDF (original Chinese and English translated version) of
Guidelines for Pharmacovigilance Inspections and the Annex –
Pharmacovigilance Inspection Check Points Table, please contact us via
online chat or email.
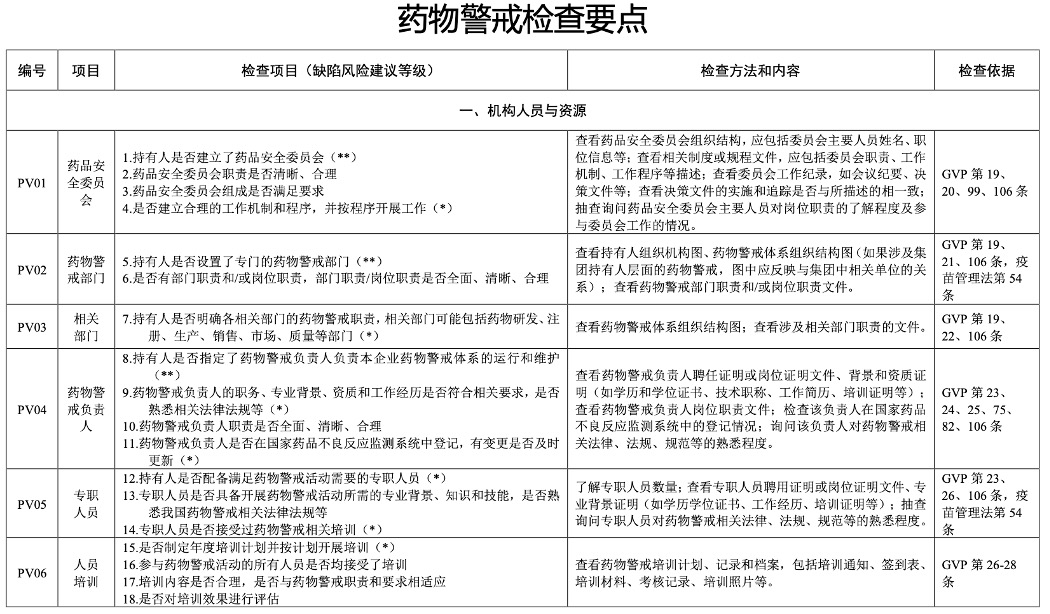
Reference:
1. Guidelines for Pharmacovigilance Inspections,
Announcement by the NMPA on the issuance of the Guidelines for
Pharmacovigilance Inspections No. [2022] 17
Literature Screening
Definition
Literature report is any adverse drug reactions reported in
- Published abstracts or
- Articles in medical/scientific journals
- Unpublished manuscripts involving case reports
- Important safety findings or clinical studies including posters,
letters to the editors, and associated communication from scientific
meetings.
Regulatory Guidance of EMA and FDA
According to the European Medicines Agency (EMA), marketing authorization
holders (MAHs) are obligated to monitor scientific and medical
publications in countries where they have marketing authorization,
regardless of the commercial status of their products.
In the United States, the Food and Drug Administration (FDA) requires the
submission of reports on serious and unexpected adverse drug reactions
(ADRs) described in scientific literature for products with the same
active ingredient as those marketed in the US, even if there are
variations in excipients, dosage forms, strengths, routes of
administration, and indications.
The “literature” section of the periodic benefit-risk evaluation report
(PBRER) necessitates a summary of significant safety findings from
published peer-reviewed scientific literature or unpublished manuscripts
during the reporting period.
EMA guidelines also mandate the inclusion of relevant safety information
for other active substances within the same class as the marketed drug.
Therefore, any potentially significant event identified in the literature
may be considered an emerging safety issue, requiring immediate analysis
and, if necessary, corrective and preventive action.
Marketing authorization holders are expected to stay informed about
possible publications through systematic literature reviews of widely used
reference databases, such as Medline, Excerpta Medica or Embase, and
Eudravigilance, at least once a week.
Each MAH should regularly screen global scientific literature using widely
used systematic reviews or reference databases, adhering to local
requirements or at least every two weeks.
Cases of ADRs reported in scientific and medical literature, including
relevant abstracts from meetings and draft manuscripts, may qualify for
expedited reporting. A regulatory reporting form with relevant medical
information should be completed for each identifiable patient, citing the
publication reference(s) as the source of the report. Additionally, the
local regulatory authority may request a copy of the article to accompany
the report. All company offices are encouraged to stay vigilant regarding
publications in local journals and inform the company’s safety department
when necessary.
The regulatory reporting timeframe begins as soon as the MAH becomes aware
that the case meets the minimum criteria for reportability.
If the specific product source, brand, or trade name is not specified, the
MAH should assume that it pertains to their product, while indicating in
the report that the specific brand was not identified.
If multiple products are mentioned in an article, a report should only be
submitted by the applicant whose product is suspected, as determined by
the article’s author.
Regulatory Guidance of China NMPA
Per the China GVP1, MAH’s responsibility relative to ICSR generated from
the literature is depicted as below:
Chapter IV Monitoring and Reporting
Section 1 Collection of Information
Article 36 The holder shall conduct periodic screening on academic
literatures, develop rational screening strategy and determine the
frequency of such screening according to the safety characteristics of the
product, etc., and the time range of the searches shall be continuous.
Chapter IV Monitoring and Reporting
Section 3 Submission of Report
Article 50 For adverse drug reactions reported in the literature, if the
suspected drug is the holder’s product, the reaction shall be reported as
an individual adverse drug reaction. If it is uncertain whether it is the
holder’s product, the reaction shall be analyzed in the periodic safety
update report and may not be reported as an individual adverse drug
reaction.
Processing of Confirmed ICSRs
- For literature reports of confirmed cases which can generate ICSRs, a
full text of the citation is obtained and, if not in English, translated
into English though it is not specified how/who does this or how long
this takes). - The article is reviewed and the number of valid ICSRs is determined, and
seriousness/non-seriousness is noted. - An ICSR is then created along with a case narrative for serious cases.
No narrative is prepared for non-serious ICSRs. - Causality assessment and relatedness also performed.
A regulatory reporting form with relevant medical information should be
provided for each identifiable patient. The regulatory reporting time
clock starts as soon as the MAH has knowledge that the case meets minimum
criteria for reportability.
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Management System
Pharmacovigilance System Master File (PSMF)
PSMF is a comprehensive document that describes the pharmacovigilance
system of a company or organization involved in the development,
manufacturing, and distribution of medicinal products. The purpose of the
PSMF is to provide regulatory authorities with an overview of the
company’s pharmacovigilance system and to demonstrate compliance with
legal requirements.
The PSMF includes information on the organizational structure, roles and
responsibilities of the personnel involved in pharmacovigilance
activities, the procedures and processes for collecting and evaluating
safety data, risk management plans, and other relevant documents related
to the company’s pharmacovigilance system. It also provides information on
deficiencies in the system and non- compliance with the requirements, the
action and measures that could be taken for specific pharmacovigilance
activities.
PSMF is a mandatory document for companies that hold a marketing
authorization for medicinal products, and it is subject to internal audits
and regulatory inspections by regulatory authorities (NMPA). In addition,
the PSMF should be updated regularly to reflect changes in the company’s
pharmacovigilance system and to ensure compliance with evolving regulatory
requirements.
In summary, the PSMF is a critical document that serves as a blueprint for
the company’s pharmacovigilance activities and provides assurance to
regulatory authorities that the company is actively monitoring the safety
of its products and taking appropriate measures to manage any risks
associated with their use.
Per the Chinese GVP1
Chapter VII Documents, Record and Data Management
Section 2 Pharmacovigilance System Master File
Article 104 The holders shall create and maintain Pharmacovigilance System
Master File to describe the pharmacovigilance system and activities.
Article 105 The holders shall update the Pharmacovigilance System Master
File in a timely manner to ensure consistency with the current
pharmacovigilance system and activities and continue to meet relevant
laws, regulations and actual work needs.
Article 106 The pharmacovigilance system master file shall include at
least the following contents:
- Organizational structure: describe the organizational structure,
responsibilities and mutual relationships related to pharmacovigilance
activities; - Basic information of the pharmacovigilance responsible person:
including residential region, contact information, resume,
responsibilities, etc.; - Staffing of full-time personnel: including the number of full-time
personnel, relevant professional backgrounds, responsibilities, etc.; - Sources of information on suspected adverse drug reactions: describe
the main channels and methods of information collection on suspected
adverse drug reactions; - IT tools or systems: describe IT tools or systems used to carry out
pharmacovigilance activities; - Management system and operating procedures: provide a brief
description of the pharmacovigilance management system, and a catalog
of the pharmacovigilance management system and operating procedures; - The operation of the pharmacovigilance system: describe the monitoring
and reporting of adverse drug reactions, and the identification,
assessment and control of drug risks; - Entrustment of pharmacovigilance activities: specify the content and
time limit of entrustment, entrusted unit, etc., and provide a list of
entrustment agreements; - Quality management: describe the quality management of
pharmacovigilance, including quality objectives, quality assurance
systems, quality control indicators, internal audits, etc.; - Appendix: Including system and operating procedural documents, drug
list, entrustment agreement, internal audit report, and master file
revision log, etc.
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Pharmacovigilance Risk Management Plan (RMP)
Pharmacovigilance Risk Management Plan (RMP) is an essential component of
the risk management strategy for a medicinal product. It is a
comprehensive document that outlines the potential risks associated with
the use of the product and the measures that will be taken to mitigate
those risks. The RMP is required to be submitted to regulatory authorities
at the time of product approval, and it is periodically updated throughout
the product lifecycle.
The RMP includes a description of the safety profile of the product,
including known and potential risks, as well as uncertainties and gaps in
knowledge. It also includes a summary of the safety data collected during
the pre-clinical and clinical development of the product. The RMP outlines
the risk minimization measures that will be implemented, including
pharmacovigilance activities, safety monitoring, and risk communication.
The RMP also includes a plan for post-authorization safety studies (PASS),
which are studies conducted after the product has been approved to further
evaluate its safety profile. The PASS may be required by regulatory
authorities as a condition of approval or as part of risk management plan
updates. The RMP outlines the objectives, design, and timelines of the
PASS.The RMP is a dynamic document that is updated throughout the product
lifecycle as new safety data becomes available or as new risks are
identified. It is an essential tool for ensuring the safety of the product
and for meeting regulatory requirements.
According to China GVP1
Chapter VI Risk Control
Section 3 Pharmacovigilance Plan
Article 96 The pharmacovigilance plan, as part of the post-marketing risk
management plan, is a written document describing the post-marketing drug
safety characteristics and how to manage drug safety risks.
Article 97 The holders shall formulate and implement a pharmacovigilance
plan based on the results of the risk assessment if important risks are
found in a marketed drug, and update the plan in a timely manner according
to changes in risk perception.
Article 98 The pharmacovigilance plan includes an overview of drug safety
and pharmacovigilance activities, and a description of the risk control
measure to be taken and the implementation time period, etc.
Article 99 The pharmacovigilance plan shall be reported to the holder’s
Drug Safety Committee for review.
Currently in China, the Post-marketing Pharmacovigilance Plan (PVP)
requirement is described and incorporated in the Guidance document for
writing Clinical RMP2. It is anticipated that in the future NMPA may
specify the Risk Control measures and activities respectively in terms of
clinical pre-phase and post-marketing phase of the products in separate
guidance documents.
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
2. Guidelines for Writing Clinical Risk Management Plans (Interim)
Announcement by the CDR of NMPA on the publication of the Guidelines for
Writing Clinical Risk Management Plans (Interim) (No. 68 of 2021)
For full PDF of Guidelines for Writing Clinical Risk Management Plans
(Pharmacovigilance Plan inclusive), please contact us via online chat or
email.
Internal Audit
See Pharmacovigilance Audit and Inspections in China (Linked to the
corresponding section upon clicking)
Entrustment Management
According to China GVP,
Chapter II Quality Management
Section 3 – Entrustment management
Article 15 Holders are the main body responsible for pharmacovigilance,
and if pharmacovigilance related work needs to be entrusted based on
business needs, the corresponding legal responsibility shall be borne by
holders.
Article 16 When holders entrust pharmacovigilance-related work to another
party, both parties shall sign an entrustment greement to ensure that the
information is true, accurate, complete and traceable, and compliant with
the requirements of relevant laws and regulations during the entire
process of pharmacovigilance activities.
Signed pharmacovigilance entrustment agreements, or written agreements can
be used between the holders of a Group and between the headquarters and
the holders, to define the corresponding responsibilities and working
mechanisms. The corresponding legal responsibilities shall be borne by the
holders.
Article 17 Holders shall examine and select entrusted parties who have
appropriate pharmacovigilance conditions and capabilities. The entrusted
parties should be legal persons within the territory of China that can
guarantee the effective operation of relevant pharmacovigilance work, have
corresponding working capabilities, and have the professional personnel,
management system, equipment resources and other working conditions to
undertake the entrusted pharmacovigilance activities. The entrusted
parties should coordinate the holder to accept the extended inspection by
the drug regulatory authorities.
Article 18 Holders shall perform periodic audit to the entrusted parties
and require the entrusted parties to fully understand the quality
objectives of their pharmacovigilance and ensure that the
pharmacovigilance activities continue to meet the requirements.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Pharmacovigilance System
The China GVP indicted the requirement that MAH are obligated when using
IT system to support the PV activities1:
Chapter III Organization Personnel and Resources
Section 3 Equipment and Resources
Article 29 The holders shall be equipped with the equipment and resources
required for pharmacovigilance activities, including office areas and
facilities, a safe and stable network environment, paper and electronic
data storage space and equipment, literature resources, medical
dictionaries, IT tools or systems, etc.
Article 30 When using an IT system to carry out pharmacovigilance
activities, the following requirements should be met:
- Define the management requirements for the design, installation,
configuration, validation, testing, training, use, maintenance and
other processes of the system, and record the above processes in a
standardized manner; - Define the security management requirements of the system, and select
control methods such as access control, authority allocation, audit
trail, authorization change, and electronic signature, etc. according
to different levels to ensure the security of the system and its data; - The system shall have complete data security and confidentiality
functions to ensure that electronic data is not damaged, lost, leaked.
Proper verification or validation shall be carried out to prove that
it meets the intended purpose.
See also Management System – PSMF
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Signal Detection
See Signal Management in China: An Overview of Regulatory Requirements
(Linked to the corresponding section upon clicking)
Aggregate Report
See Aggregate Reporting in China (Linked to the corresponding section upon
clicking)
Post-Authorisation Safety Study (PASS)
According China GVP1,
Article 71 The holders shall take the initiative to carry out drug
post-marketing safety studies based on the risk of the drug, or carry out
drug post-marketing safety studies in accordance with the requirements of
the provincial or higher drug regulatory authority. Post-marketing safety
studies and activities of drugs shall not be aimed at product promotion.
Article 74 The holders shall select appropriate post-marketing safety
study methods based on the study objectives, drug risk characteristics,
clinical use, etc. Post-marketing safety studies of drugs can be based on
primary data collected directly from medical staff or patients in this
study, or based on secondary data that has occurred before this study and
been collected for other research purposes.
Article 75 The holders shall formulate a written study protocol for
post-marketing safety studies. The study protocol shall be formulated by
personnel with appropriate academic background and practical experience,
and reviewed and approved by the pharmacovigilance responsible person.
The study protocol shall stipulate the procedures for the collection,
assessment and reporting of the information on suspected adverse drug
reactions during the study period, and shall be summarized in the study
report.
During the study process, the study protocol can be revised or updated as
needed. After the start of the study, any substantial revisions to the
study protocol (such as study endpoints and study population changes)
shall be recorded in the protocol in a traceable and reviewable manner,
including the reason, content, and date of the change.
Article 76 For drug post-marketing safety studies required by the drug
regulatory authority, the study protocol and report shall be submitted
according to the requirements of the drug regulatory authority.
Article 77 The holders shall monitor the safety information during the
study period, and when discovering any new information that may affect the
benefit-risk balance of the drug, it shall conduct an assessment in a
timely manner.
An official response to Recommendation No. 9017 of the Fifth Session of
the 13th National People’s Congress of NMPA explained the current
challenges that have been encountered2:
In order to guide holders to standardize the conduct of post-marketing
safety studies on drugs, NMPA issued the GVP in May 2021. The GVP fully
draws on international regulatory experience regarding post-marketing
safety studies of drugs, and clarifies the scope, purpose, ethical
requirements, research methods, data sources, research protocols, research
reports and other ten normative requirements for holders to conduct
post-marketing safety studies of drugs, so that holders have rules to
follow in conducting post-marketing safety studies of drugs. Since the
release of the GVP, NMPA has organised several training sessions to
disseminate and guide the holders and continuously promote them to improve
the capability and level of post-marketing safety studies.
In the next step, the NMPA will continue to support the research and
development of innovative drugs driven by clinical value, and continue to
improve the relevant working mechanism in collaboration with relevant
departments to better meet public demand for medicines.
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
2. Response to Recommendation No. 9017 of the Fifth Session of the 13th
National People’s Congress
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Comparison Between China PV and Europe PV
Comparison of China GVP and EMA GVP

The European Medicines Agency (EMA) has developed 12 modules (I to XVI)
for pharmacovigilance processes. The EMA has already covered the topics
for Modules XI to XIV in other guidance documents, and thus they are null.
The remaining modules play a vital role in ensuring patient safety.
Module I focuses on quality objectives for pharmacovigilance systems to
comply with safety standards. It guides pharmaceutical companies on how to
handle each aspect of the process to meet those objectives.
Pharmacovigilance systems are used to identify if a drug has adverse
effects and to track drug safety over time. Organizations can show their
findings to regulatory authorities and alert the public if new adverse
events are detected and verified.
Module II requires the inclusion of a pharmacovigilance system master file
(PSMF) with every marketing authorization (MA) application. The PSMF must
contain details on the Qualified person responsible for pharmacovigilance,
organizational structure, computer system, PV processes, quality system
activities, delegated activities.
Module III and IV are pharmacovigilance inspections and audits
respectively, which are conducted by competent authorities to ensure
compliance with EMA guidelines. Inspections ensure the availability of
resources to meet pharmacovigilance requirements, while audits verify the
ability of pharmacovigilance systems to perform activities effectively.
Module V involves the creation of a risk management plan (RMP) for every
drug. The RMP identifies the safety profile of the drug and creates a
pharmacovigilance plan to identify new adverse reactions. As more
information emerges, the RMP adapts to changes in the drug’s safety
profile.
Module VI provides guidance on the collection, management, and submission
of reports of suspected adverse reactions to medicinal products. The
reports are collected by pharmacovigilance systems and evaluated by
competent authorities and MA holders.
Module VII focuses on Periodic Safety Update Reports (PSURs) which provide
an overview of a drug’s risk-benefit during the post-authorization phase.
The competent authorities review the PSURs within 70-90 days of the data
lock point to check if the risk-benefit balance has changed.
Module VIII involves conducting Post-Authorization Safety Studies (PASS)
to identify a safety hazard or confirm the safety profile of a drug. These
studies are initiated by the EMA or the MA holder and reviewed by the
Pharmacovigilance Risk Assessment Committee (PRAC).
Module IX refers to signal management which is about the evaluation,
reporting, and timelines of drug safety issues. Pharmaceutical companies
need to send a signal to the EMA within 30 days of identifying an adverse
reaction.
Module X involves additional monitoring for certain drugs that may have
adverse reactions emerge after authorization. The EMA maintains a list of
these drugs which require additional data collection, and they are
identified with an inverted equilateral black triangle ▼.
Module XV is about safety communication, and it involves disseminating
information to healthcare professionals and the public about the safe use
of drugs.
Module XVI focuses on the implementation of risk minimization measures to
minimize risks associated with drug use. These measures are based on the
RMP and are regularly reviewed and updated. Overall, these modules help in
ensuring the safety of patients and the public.
China GVP on the other hand showed similar structures. China GVP is based
on the “Guidelines for Adverse Drug Reaction Reporting and Monitoring”
issued by the Chinese Ministry of Health, while EU GVP is developed by the
European Medicines Agency (EMA) in collaboration with the EU member
states.
In terms of content, both frameworks cover similar aspects as illustrated
in the graph with same colour highlighted for the PV aspects covered.
However, there may be differences in specific requirements and guidelines
due to variations in regulatory processes and regional considerations. It
is important for pharmaceutical companies operating in both regions to be
familiar with and adhere to the respective GVP guidelines to ensure
compliance with local pharmacovigilance requirements.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
China PSMF vs EMA PSMF

The establishment of China PSMF referenced EMA GVP Module II of PSMF as a
model. Nevertheless, two additional aspects “Entrustment of
Pharmacovigilance Activities” and “Full-time Personnel” are incorporated
in the China PSMF as required in the Guideline for Preparation of the
Pharmacovigilance System Master File published by NMPA.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Responsible Person for Pharmacovigilance – A Thorough Analysis of Current
China “QPPV” System
The implementation of Pharmacovigilance activities in China is governed by
the China (GVP), which mandates Marketing Authorization Holders (MAHs) to
establish pharmacovigilance quality objectives and a comprehensive quality
assurance system. To fulfill PV responsibilities, MAHs are required to
introduce the concept of the Qualified Person for Pharmacovigilance
(QPPV). Similarly, the European Union requires MAHs to establish a
Pharmacovigilance system and appoint a QPPV. However, the United States
does not have explicit requirements for Pharmacovigilance systems.
The official implementation of the China GVP is on December 1, 2021,
emphasising the primary responsibilities of MAHs in Pharmacovigilance.
Despite the existence of Level 3 Adverse Drug Reaction Monitoring Centres,
a shortage of QPPVs has been observed in recent years in China.
The qualifications and responsibilities of QPPVs within the GVP quality
management system have led to variations and confusion within MAHs. This
includes differences in position designation, hierarchical level, duties,
and the required knowledge and skills for QPPVs. The QPPV plays a vital
role as the main contact between the MAH and Regulatory Authorities,
ensuring drug safety and compliance with regulations through the
establishment and maintenance of pharmacovigilance systems, monitoring and
reporting adverse drug reactions, and implementing risk management
strategies.
Analysis of QPPV Position Requirements in China – General Requirements for
QPPV Qualifications
In China GVP it is required that the QPPV “should be a managerial position
with certain responsibilities, have a background in medicine, pharmacy,
epidemiology, or related fields, possess a bachelor’s degree or higher
education, or have an intermediate or higher professional and technical
title.” It also states that the QPPV should have “over three years of
experience in pharmacovigilance-related work, be familiar with
Pharmacovigilance-related laws and regulations and technical guidance
principles in China, and have the knowledge and skills for
Pharmacovigilance management”.
The selection requirements of EU QPPV also include similar professional
background requirements, with a specific emphasis on medical knowledge or
a background in medicine. The EU-GVP mentions that the EU-QPPV should
possess the skills to manage the pharmacovigilance system and have the
expertise or ability to acquire knowledge in relevant fields such as
medicine, pharmacy, epidemiology, and biostatistics.
Analysis of QPPV Position Requirements in China – Professional and
Technical Responsibilities of QPPV
In China GVP, it is required that the QPPV should have “over three years
of experience in pharmacovigilance-related work,” meaning that they have
been dedicated to working in drug vigilance and have experience in
activities related to the collection and reporting of drug safety
information (such as physicians, pharmacists, CRA/CRC personnel in
clinical research), as well as adverse drug reaction (ADR) handling.
Similarly, in the EU-GVP, EU-QPPV applicants or MAHs are subject to
qualification review and assessment before appointment, including
evaluation of university education, understanding of EU pharmacovigilance
requirements, and experience in pharmacovigilance.
According to Article 25 of the China GVP, the QPPV is responsible for the
operation and continuous improvement of the pharmacovigilance system,
ensuring its compliance with relevant laws, regulations, and the
requirements of the GVP. Therefore, the responsibilities of the QPPV can
be summarized as continuous improvement, compliance, risk control,
information communication, and auditing.
- The foundation of pharmacovigilance quality management work is based
on the pharmacovigilance system as the main focus. The QPPV implements
continuous improvement plans through the Deming cycle (Plan, Do,
Check, Act), ensuring the effective operation of the pharmacovigilance
system. - Compliance of monitoring reports is one of the quality objectives of
pharmacovigilance. By setting quantifiable quality control indicators,
the effectiveness of the pharmacovigilance system is measured. If the
compliance indicators are not met, the QPPV should lead the analysis
of errors, training, processes, and other reasons. - Pharmacovigilance is work based on the principle of “full-process
control and risk management.” The QPPV should supervise the entire
process of pharmacovigilance, conduct drug safety risk identification,
assessment, and control, and ultimately ensure the effective
implementation of risk control measures. - Channels of communication in pharmacovigilance should remain open,
including multiple channels for reporting safety information (phone,
fax, email, mobile applications, etc.). QPPV should be contacted
(department) promptly through convenient means to provide feedback on
safety issues or other work instructions, such as conducting
inspections and benefit-risk assessments, and respond adequately and
timely to inquiries from Regulatory Authorities. - Safety information is one of the important contents of
pharmacovigilance work. The QPPV is responsible for the accurate and
complete communication and transmission of information to ensure
timely and effective communication. - The QPPV needs to review and issue important documents, including
periodic safety update reports and post-marketing safety study
protocols. These documents reflect the integrity of the
pharmacovigilance system or the safety of specific products. However,
it is not recommended to include Individual Case Safety Report files
in the category of important documents.
Comparison of QPPV Positions between China and the European Union –
Qualifications for Appointment
There are some differences in the qualifications for QPPV positions
between China and EU, mainly in terms of experience, knowledge and skills,
and personnel attributes (see Table 1).
- Education and professional background: No significant differences
between China and EU. Both require a Bachelor degree or above, with a
focus on relevant fields such as medicine and pharmacy. However, the
EU places more emphasis on the field of biostatistics. Due to the
interdisciplinary nature of Pharmacovigilance, both China and the EU
face a shortage of professionals in the field, allowing individuals
with relevant backgrounds to assume QPPV responsibilities. - Experience and qualifications of QPPV: China specifies a requirement
of 3 years of relevant experience in pharmacovigilance, while the EU
does not explicitly mention the years of experience. - Knowledge and skills: Both China and the EU require familiarity with
local pharmacovigilance-related laws, regulations, and technical
specifications. In China, as the initial version of GVP was just
released in May 2021, it may take some time for QPPVs to acquire the
necessary knowledge in the field of drug vigilance. Therefore, if
Chinese pharmaceutical companies choose to hire or outsource QPPV
positions, it may be possible to quickly meet legaland policy
requirements. - Duties and place of residence: China requires QPPVs to hold management
positions and expects them to influence relevant personnel and
departments through these positions, which is more conducive to
conducting Pharmacovigilance work. The EU-GVP does not specify
management position requirements, although it emphasizes
responsibilities and influence, which typically requires time
accumulation in managerial roles. The EU also emphasizes that QPPVs
should reside within the EU or in Norway, Iceland, or Liechtenstein.
China does not mention residence requirements, but in general, it is
preferable for QPPVs to be located within China, as it facilitates
communication with Regulatory Authorities. - Personnel attributes of QPPV: The Chinese GVP does not mention the
outsourcing or employment of external QPPV personnel, while the EU-GVP
allows QPPVs to be employees of third-party companies to oversee and
manage the pharmacovigilance system of MAHs.
QualificationChina RPPVEU-QPPV
| Educational Backgraound | Bachelor or Mid-level and above Technical Titles | University/College |
| Major | Medicine, Pharmacy, Epidemiology or related majors | Medicine, Pharmacy, Epidemiology, Biostatistics and others |
| Knowledge/Skill | Familiar with China’s pharmacovigilance-related laws and regulations and technical guidelines, with knowledge and skills in pharmacovigilance management |
Familiarity with EU pharmacovigilance requirements |
| Experience | At least three years of experience in pharmacovigilance related work | Experience in pharmacovigilance required |
| Roles | Managerial positions | May be an independent individual, not mentioned as requiring a managerial position |
| Residence | not mentioned, generally within China | Norway, Iceland, or Liechtenstein |
| Personnel Attibute | Not mentioned if non-MAH employee is allowed | MAH Employee or Entrusted |
Table 1 Comparison of QPPV Qualification between China and EU.
Comparison of QPPV Positions between China and the European Union – Job
Responsibilities
There are differences in job responsibilities between QPPV positions in
China and the European Union, including compliance management,
communication management, government-enterprise communication, as well as
management and legal responsibilities. The initial version of GVP in China
provides a relatively concise and clear description of QPPV
responsibilities. In Article 25, under “Personnel and Training,” it
briefly outlines the responsibilities of QPPV, such as
compliance,supervision, communication management, government-enterprise
communication, and review and issuance. The Pharmacovigilance System
Master File (PSMF) also requires basic information about the QPPV,
including residence area, contact information, resume, and
responsibilities. On the other hand, the European Union’s GVP provides a
more detailed introduction to the responsibilities and role of the QPPV.
The supervisory responsibilities include standard operating procedures,
contract arrangements, database operations, compliance data related to
quality, regular updates and auditing reports, completeness and timeliness
of expedited reporting, training on pharmacovigilance, and contracted
management. Please refer to Table 2 for more details.
Regarding the management responsibilities of the PSMF, the Chinese GVP
does not mention or emphasize the QPPV’s authority over PSMF management or
specify the personnel responsible for reviewing and issuing the PSMF.
However, as a system description document, the European Union particularly
emphasizes the QPPV’s management authority over the PSMF. In terms of
legal responsibilities, there are differences between China and the
European Union in terms of specific legal obligations. In China, the QPPV
is primarily responsible for technical guidance, specifically for the
technical aspects of drug vigilance,without mentioning the need to assume
corresponding legal responsibilities and risks. On the other hand, the
European Union QPPV has legal responsibilities, meaning that the position
carries certain risks, and it is necessary to ensure compliance with legal
requirements to avoid personal and company penalties. Therefore, based on
the practical experience of QPPVs in the European Union in terms of
quality improvement, management responsibilities, and legal regulations,
as well as the early stage of establishing the drug vigilance system in
China, it is necessary for China to develop more detailed specifications
for QPPV job responsibilities. These specifications can be used by
pharmaceutical companies and MAHs to implement QPPV selection from the
perspectives of risk management and compliance management.
Outsourcing Analysis of QPPV – Current Human Resources Situation of QPPV
in China
Currently, there are no specific laws and guidelines in China that
explicitly state whether MAHs can outsource QPPV or whether QPPV should
assume legal responsibilities. Possible reasons for this could be based on
the experience of implementing Good Manufacturing Practices (GMP) and Good
Supply Practices (GSP), where key personnel responsible for quality
management should be full-time employees of the company, without
precedents for outsourcing or hiring third-party personnel. At the same
time, considering the current status of practicing pharmacists and Risk
Management of MAH in China, no sufficient QPPVs who truly meet regulatory
requirements to meet market demand. It may also be considered that most
companies can initially meet transitional standards, and regulations and
norms only explicitly state that the entity’s responsibility lies with the
legal entity or the primary person in charge, without requiring the QPPV
to assume legal responsibilities.
When implementing outsourcing for a company’s pharmacovigilance program,
it is possible to consider engaging third-party personnel to assume the
responsibilities of QPPV on behalf of the MAH. In this case, there is no
labor contract between the MAH and the QPPV, but rather a service contract
that binds them. This method of outsourcing QPPV hiring can draw lessons
from the QPPV system in the European Union.
Outsourcing Analysis of QPPV – Relevant Regulations on Pharmacovigilance
Agreements
With the advancement of GVP learning and exchange both domestically and
internationally, as well as the gradual maturity of MAH outsourcing
business, QPPV entrusting may become an important component of MAH’s
quality system outsourcing. In the second chapter of China’s GVP, titled
“Entrustment Management,” MAH is required to “entrust
pharmacovigilance-related work based on job needs,” but responsibilities
are not transferred, and “corresponding legal responsibilities are borne
by the holder.” In 2020, the National Medical Products Administration
issued the “Guidelines for Writing Pharmacovigilance Agreements (Trial),”
but the document does not mention entrustment related to QPPV.
Reference:
1. Discussion and Thoughts on the Job Responsibilities and Selection of
QPPV between China and EU, XU Juping, HU Jun, WAN Bangxi, WEI Xiaofei,
WANG Guangping
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Individual Safety Case Report
NMPA
Reporting timelines:
- Fatal ADRs: immediately (For cases received during the weekend, submit
within next business day, no later than Day 3) - Other serious ADRs: within 15 calendar days
- Non-serious ADRs: within 30 calendar days.
EMA
Submit the valid ICSR (EEA and non-EEA serious within 15 days, and EEA
non-serious ICSR within 90 days to EudraVigilance (EV)). Non-serious
non-EEA ICSRs should not be submitted to EV.
FDA
Must report adverse drug experience that is both serious and unexpected,
whether foreign or domestic, as soon as possible but no later than 15
calendar days from initial receipt of the information and must submit
follow-up reports within 15 calendar days of receipt of new information by
the applicant.
The applicant must report each adverse drug experience involving serious
listed, non-serious unlisted and listed events at quarterly intervals, for
3 years from the date of approval of the application, and then at annual
intervals. The applicant must submit each quarterly report within 30 days
of the close ofthe quarter (the first quarter beginning on the date of
approval of the application) and each annual report within 60 days of the
anniversary date of approval of the application. Follow-up information to
adverse drug experiences submitted in a periodic report may be submitted
in the next periodic report.
MAH Responsibility
MAH responsibilities elucidated in China GVP
China GVP1 predominantly emphasizes the responsibilities of the Marketing
Authorization Holder (MAH), as a substantial portion of its articles are
dedicated to the comprehensive obligations.
Reference:
1. China Good Pharmacovigilance Practice
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
For full China GVP document, please contact us via online chat or
email.</strong >
Local Representative of Overseas MAH
The NMPA published Interim Provisions for the Administration of Local
Representatives of Overseas Marketing Authorisation Holder1 as the
guidance provision for Local Representative of Overseas MAH
Basic requirements for becoming a MAH local representative
Based on the relevant requirements of the Drug Administration Law and the
Interim Provisions,we have sorted out the two basic requirements for
foreign holders to appoint domestic agents: corporate legal person and
legal responsibility.
- An enterprise legal person established in China. It can be a subsidiary,
but not a branch, not a branch of a foreign company set up in China. - Legal liability: Article 136 of the Drug Administration Law If the
holder of a drug marketing licence is an overseas enterprise, and its
designated enterprise legal person in China fails to fulfil the relevant
obligations in accordance with the provisions of this Law, the
provisions of this Law concerning the legal liability of the holder of
the drug marketing licence shall apply: i.e. civil liability,
administrative liability, criminal liability, etc.
Liability as a MAH local representative
The new Drug Administration Law and the Interim Provisions further clarify
the responsibility for quality and safety throughout the life cycle of a
drug. The liability is borne by the MAH local representative
At present, large multinational pharmaceutical companies have set up their
head offices outside of China, and one or more branch companies are
usually responsible for registration, commercial representation, and
pharmacovigilance representation in China, while small and medium-sized
overseas pharmaceutical companies usually perform the above-mentioned
duties through pharmaceutical business enterprises in China. According to
the “Drug Administration Law” and the “Interim Provisions” requirements,
the overseas MAH can only appoint a representative to perform its duties,
which requires the domestic agent for registration, production,
distribution, quality, customs, pharmacovigilance and a series of
performance of the integration, and its responsibility for the entire
process in the territory.
The relationship between the two is reflected in the obligations to be
performed by the ” MAH local representative ”
1. Responsible for establishing a drug quality assurance system to ensure
that it has continuous quality assurance and risk control capabilities.
Enterprises need to develop a quality assurance system document that meets
their business needs and can be effectively implemented to cover the
entire life cycle of the agent’s medicines.
2. Responsible for establishing and implementing a drug traceability
system and providing traceability information in accordance with
regulations to ensure that the whole process of the relevant listed drugs
can be traced. The regulatory authority has made it clear in 2018 that it
will take the lead in establishing a drug information traceability system
for key varieties such as vaccines, narcotic drugs, psychotropic drugs,
easily controlled chemical drugs and blood products, and has pointed out
in 2020 that it will basically achieve traceability of key varieties by 31
December 2020. Domestic agents are required to establish a drug
traceability system, and according to the general principle of “one item,
one code, one code, one trace”, they can build their own traceability
system or commission a third-party technical institution to build one, and
assign a unique traceability mark to all levels of drug packaging units in
accordance with the unified drug traceability code requirements.
For overseas MAHs, how and where to assign the code, the assignment of the
code to the overseas production plant or the assignment of the code in the
free trade zone, the printing requirements of the traceability code and
the display of the traceability code consumer query results are topics
that need to be discussed separately.
3. responsible for the establishment and can implement the drug annual
report system, on behalf of the overseas holder will be confirmed by the
overseas holder each year after the relevant drug production and sales in
China, post-marketing research evaluation, risk management and other
information in accordance with the provisions of the provincial drug
supervision and management department where the agent is registered. The
NMPA has recently issued the Regulations on the Administration of Annual
Reports of Pharmaceutical Products, in which it is clarified that “if the
holder is an overseas enterprise, the enterprise legal person (domestic
agent) designated by it in accordance with the law to assume joint and
several responsibilities in China shall perform the annual reporting
obligations”. At present, the State does not specify the agency filing
requirements for domestic agents, but the Regulations on the
Administration of Annual Reports of Pharmaceutical Products show that the
holder of an overseas listing permit can designate a domestic corporate
legal person to fill in the information, and the corporate legal person
has to bind the drugs held by the overseas listing permit holder, and a
drug can only be bound by one domestic agent, and if there is a change in
the agency, the original corporate legal person has to unbind the drug and
unbind the new domestic agent. If there is a change in the agency
practice, the original corporate entity will have to unbind the medicine
and the new MAH Local representative will be re-bound. The basis for the
binding is the submission of proof of agency status by the MAH Local
representative, such as the enterprise information and power of attorney
of the unit.
4. Responsible for the establishment of a post-marketing change management
system for pharmaceutical products and for handling changes in strict
accordance with the regulations. In January 2021, the S The NMPA issued
the “Measures for the Administration of Post-marketing Changes to
Medicines (for Trial Implementation)”. In the requirements for reporting
information, it can be understood that if a drug is manufactured outside
China and the overseas holder appoints an enterprise in China to act as
MAH local representative for the relevant drug registration matters, it
should provide a power of attorney and notarized and certified documents
with Chinese translation, as well as a copy of the business license of the
registered representative in China.
5. Undertake matters such as post-marketing recall, quality complaint
handling and quality compensation of drugs, and report to the provincial
drug supervision department where the representative is registered as
required. According to the Measures for the Administration of Drug Recalls
(Draft for Public Comments), which was again made available for public
consultation in 2021, in relation to the recall of drugs manufactured
outside China involving implementation within the country, the designated
enterprise legal person within China shall organise the implementation in
accordance with the provisions of the Measures. In the case of a drug
recall implemented outside China only that does not involve a domestic
drug variety or batch, the designated corporate entity in China will
report the recall information to the drug regulatory authority. And the
designated domestic corporate entity will carry out the website
publication of the recall information and act as the recipient of the
service of the recall order notice.
6. It is responsible for establishing a pharmacovigilance system,
formulating a post-marketing risk management plan for drugs, and carrying
out monitoring, identification, assessment and control of post-marketing
adverse drug reactions and other harmful reactions related to drug use as
required. MAH local representative is required to establish and carry out
pharmacovigilance quality systems and activities within their enterprises
in accordance with the Pharmacovigilance Quality Management Code and the
Pharmacovigilance Inspection Guidelines. As a domestic agent, you can
carry out pharmacovigilance work on your own or commission
pharmacovigilance-related work according to the actual situation of your
company. It is required to have the ability to collect information on
drugs outside of China and to promptly report the suspension of sales, use
or withdrawal of drugs from the market due to safety reasons.
7. Responsible for submitting standard substances to the China Academy of
Food and Drug Administration in accordance with relevant regulations, and
accepting sampling tests organised and carried out by the drug regulatory
authorities. When the drug agent accepts the sampling and testing
requirements of the drug regulatory authorities, the domestic agent is
responsible for submitting the standard substances and cooperating with
the relevant work.
8、Responsible for liaising with the overseas holder and cooperating with
the drug regulatory authorities to carry out inspections, investigations
and investigation of violations of the law at the production site of the
overseas holder. In summary, as a domestic agent, there will be multiple
identities in the act of representation, which may be both the customs
service provider of the represented pharmaceutical product, the domestic
sub-package manufacturer of the represented pharmaceutical product, or its
domestic business distribution enterprise. Regardless of the identity, as
a domestic agent itself, you need to have comprehensive quality assurance
capabilities, as well as the ability to perform under different regulatory
areas, and even if you outsource some of the functions, you need to have
the ability to manage the third party entrusted to you in order to control
the risks and achieve mutual benefits for both parties.
Reference:
1. Public consultation request for “Interim Provisions on the
Administration of Local Representative for Overseas Marketing
Authorisation Holder” by the General Department of the NMPA
2. https://www.ciopharma.com/article/explain/497
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
PV Safety Databases
ArisG Safety Database
ArisG1 uses artificial intelligence to automate case processing,
facilitate decision-making and make safety workflows more efficient. It is
a cloud-based solution that allows for easy updating to new regulatory
standards and global PV teams to operate in one real-time system. You can
visualize reporting measures and quickly generate compliance and local or
regional regulatory reports. The safety database can be configured to
individual client’s needs, the medicinal product(s), studies, reporting
requirements etc.
features include:
- Global cloud-based access with real-time review of cases
- Automated coding
- Automated duplicate checks
- Automated validity and triage of cases
- Configuration of client-based SOPs, workflows, review and reporting
procedures - Integration with other PV systems, such as safety signal management
- Automated case tracking and follow-up
- Real-time benefit-risk analysis
- Global automated submission options
Argus Safety Databases
Argus Safety is a comprehensive global safety database that leverages
automation to streamline case processing and generate in-depth analytics
and reporting. The platform is highly customizable and can be tailored to
meet the unique needs of each client, including dynamic workflows and
specific reporting rules. It can also integrate seamlessly with
third-party or internal pharmacovigilance systems. By providing automated
distribution and submission of reports, Argus Safety ensures regulatory
compliance through the configuration of specific rules appropriate to the
respective medicinal product(s) and offers reporting and compliance
metrics to its users. Additionally, features such as automated reporting,
document storage, and integration with safety signal detection and
management make it an essential pharmacovigilance safety database. The
ability to update the drug safety profile and integrate it into risk
management decision-making further enhances its value. Other key features
of Argus Safety include worldwide cloud-based access, automation-assisted
case processing, duplicate control, case tracking, configuration for
regional regulatory requirements, client-based rules for SOPs, workflows,
review and reporting procedures, automated submission options, options for
integration with other PV processes, and audit trails to ensure data
accuracy and compliance.
Chinese Safety Databases
Regarding Chinese Local Safety Databases, sophisticated supplier like
TAIMEI is the most popular on the market due to its strength in
localization of electronic transmission with China local regulatory
authories and facilitation of communication channels. However the local
supplier may also experience challenges and encounter limitations on
providing the Exhaustive resolution to meet the regulatory requirement.
Accestra offer consultation with evaluation to recommend the best
system/databases tailored to your company.
Click here to learn more
(NOTE: once clicked Click here, it will direct to the Accestra contact
page/pop up window)
Reference:
1.
https://www.biomapas.com/selecting-a-pharmacovigilance-drug-safety-database/</a >
2. https://www.arisglobal.com/</a >
3.
https://docs.oracle.com/en/industries/health-sciences/argus-safety/index.html</a >
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
FAQs
Case Reporting (ICSR Reporting)
What are the requirements for reporting overseas adverse drug reactions
(ADRs) in pharmacovigilance?</strong >
According to the Provision for Adverse Drug Reaction Reporting and
Monitoring (Order No.81 of the Ministry of Health), for Serious adverse
drug reactions (including those collected by the spontaneous reporting
system, those found in post-marketing clinical studies and those reported
in the literature) occurring overseas for both imported and domestic drugs
shall be reported to the National Adverse Drug Reaction Monitoring Centre
(NADRMC) within 30 days from the date of notification. If the National
Adverse Drug Reaction Monitoring Centre requires the original report and
related information, the MAH shall submit it within 5 days.
Should you have any queries or seeking for consultation, please contact
us via online chat or email.
Responsible Person for Pharmacovigilance (RPPV) in China
Who can serve as a RPPV in China?
According to China GVP,
Article 24 The pharmacovigilance responsible person shall be in certain
managerial positions, have a medical, pharmaceutical, epidemiology or
relevant professional background with a bachelor degree and above or with
a professional technical title of mid-at level and above, have an
experience in pharmacovigilance-related work for more than 3 years, be
familiar with China pharmacovigilance-related laws, regulations and
technical guidelines, and have the knowledge and skills to manage
pharmacovigilance work.
The pharmacovigilance responsible person shall be registered in the
National Adverse Drug Reaction Monitoring System. If the relevant
information is changed, the pharmacovigilance responsible person shall
complete the update within 30 days from the date of the change.
What are the responsibilities of a RPPV in China?
Per China GVP Article 25, The pharmacovigilance responsible person is
responsible for the operation and continuous improvement of the
pharmacovigilance system, ensuring that the pharmacovigilance system meets
the requirements of relevant laws, regulations, and this practice. The
main responsibilities of this person include:
- To ensure compliance of adverse drug reactions monitoring and
reporting; - To oversight the identification, assessment and control of drug safety
risks and ensure the effective implementation of risk control
measures; - To be responsible for the management of drug safety information
communication and ensure timely and effective communication; - To ensure smooth communication channels within the holders and with
the drug regulatory authorities and the drug adverse reaction
monitoring authorities; - To be responsible for the review or sign-off of important
pharmacovigilance documents.
Normally the transnational corporations have global PV system, so can
the QPPV from headquarter play the role of QPPV in China and does China
NMPA require QPPV shall be reachable 24 hours?</strong >
In China, it’s called Responsible Person for Pharmacovigilance, which
equals to QPPV. There is no specific requirement that only persons based
in China can take the role of RPPV. But practically speaking, it is
recommended to assign a person based in China to be local RPPV and keep
contact available always so that authorities can reach the RPPV when
necessary.
Can the leader of PV or QA Department play the role of QPPV?</strong >
According to NMPA experts, it is not suggested to assign department
managers, like PV or QA department, to be RPPV/QPPV because it’s required
that the lead of PV shall have enough power to control PV activities and
make decision in the company, like vice president. However, every company
may have different situation to appoint the RPPV, but the key is the
continuous training on the appointed person and RPPV is not just a role
with title.
Reference:
1. China Good Pharmacovigilance Practice (GVP)
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021)
Pharmacovigilance Inspection
How is the inspection performed?
China National Medical Products Administration (NMPA) is the leader who
designate or set up the inspection institutions and assign inspectors to
support local MPAs. With practical assistance of provincial MPA, they
build inspector information platform together and then assign inspection
team to actually conduct the on-site inspection in pharma companies. The
lower tiers of MPA can apply inspector supports from the higher authority.
The inspection team is consisted of inspectors from centers for drug
control/testing, evaluation for approval, reevaluation, and ADR monitoring
of national or local authorities. If there are any unlawful cases, then
the legislative affairs office may be involved in and assist with the case
handling.
Pharmacovigilance Safety Update Report (PSUR)
What is the PSUR submission frequency in China?
Per the Periodic Safety Update Reports Writing Guidelines1, the data for
the Periodic Safety Update Report shall be summarised from the date of
obtaining the drug approval document and shall be reported within 60 days
of the data cut-off date. It is possible to submit a Periodic Safety
Update Report starting from the international date of birth, but if the
data cut-off date for the above-mentioned report is earlier than the
cut-off date required in China, the data for this period should be
supplemented and analysed.
Reference:
1. Guidance for Writing Periodic Safety Update Reports of Drugs
Announcement of the National Food and Drug Administration (NFDA) on the
Issuance of Guidance for Writing Periodic Safety Update Reports of Drugs
(No.[2012]264)
Can MAH submit PBRER instead of PSUR and what’s the specific
requirements?</strong >
Yes, MAH can use PBRER to replace PSUR. According to the announcement
(NMPA No. 2020/86) released in July 2020,PBRER can replace PSUR, and the
format and submission deadline shall follow ICH E2C (R2) Periodic
Benefits-Risks Evaluation Report, which means PBRER with reporting period
no more than one year shall be submitted within 70 days since DLP and
PBRER with reporting period more than one year shall be submitted within
90 days since DLP.
Which part of imported drug’s PSUR shall be translated into Chinese
before submission?</strong >
Except for Line Listings and Summary Tabulations, all the other parts of
PSUR shall be translated into Chinese and submitted with CCDS (Company
Core Data Sheet) and original English version of PSUR.
Which date shall be the starting date for PSUR submission? If the
submission is overdue, will PSUR be rejected by the system?</strong >
Usually globally speaking, it starts from IBD, but if no information about
IBD then CBD (approval date in China).If you miss the deadline of PSUR
submission, the National ADR Monitoring System will still accept the
submission but it does not mean the report passes.
Pharmacovigilance System Master File (PSMF)
Is it feasible to convert EMA PSMF to China version of PSMF?</strong >
Since the design of China PSMF is referencing EMA PSMF template, there is
plenty of similarity in terms of structures of the Master File. it is
feasible to convert the required elements into the Chinese version of PSMF
suitable for China Regulatory Authorities. Contact us for our PV expert
for PSMF conversion consultation.
Drug Safety Committee (DSC)
or transnational pharma corporations, can the drug safety committee
from headquarter take the role for China?</strong >
It depends. Generally the Drug Safety Committee concerns the drug safety
globally from an overall point of view, but some markets may have
different features and requirements. Furthermore, according to article 20
of GVP, the DSC is responsible for significant risk’s judgement &
determination, significant or emergency events’solution, risk control
decision-making and other important events related with pharmacovigilance.
Significant events are normally very urgent and it is required to respond
immediately accordingly. But HQ Safety Committee may not even have enough
time to respond for emergencies. So, it would be better to have local
Safety Committee. However, if the HQ can make sure that they can respond
timely and the SOP is compliant with China requirement, it’s also
feasible.
Risk Management Plan (RMP)
Shall all drugs create the RMP?
Not all kinds of drugs are mandatory to create a RMP or PV Plan according
to regulations. It is required for those marketed drugs found with
significant risks according to risk assessment result and to keep the plan
updated.
Shall we make RMP and PV Plan both or alternative?
The PV Plan can be used as RMP and vise versa. For instance, if you
already have a pre-marketing RMP, you can adjust it into a PV Plan or a
post-marketing RMP accordingly.
Does the Regulatory Authority release the official Post-marketing
Pharmacovigilance Plan Guideline document?</strong >
Currently the regulatory requirement for PVP is included in the Clinical
Risk Management Plan Writing Guideline document.
Safety Database in China
What is the regulatory requirement for safety databases in
China?</strong >
In China, it is mandatory for pharmaceutical companies to establish a
safety database to collect and manage adverse drug reaction (ADR) reports
as per the China GVP1 and ADR Reporting Guideline2 released by the
National Medical Products Administration (NMPA).
Can the safety database in China be outsourced to a third-party service
provider?</strong >
Yes, outsourcing the safety database function to a third-party service
provider is allowed in China, but the pharmaceutical company must ensure
that the provider has adequate experience, expertise, and a robust quality
management system.
Reference:
- China Good Pharmacovigilance Practice (GVP)
Announcement of the NMPA on the Publication of China Good
Pharmacovigilance Practice (No. 65 of 2021) - Guidelines for the Collection and Reporting of Individual Adverse Drug
Reactions
Announcement by the NMPA on the Issuance of Guidelines for the
Collection and Reporting of Individual Adverse Drug Reactions (No.
[2018] 131)