Individual Adverse Drug Reactions Reporting and Monitoring in China
What is an Individual Case Safety Report (ICSR) in China?
So, what is an ICSR? According to the ICH E2 series of related guideline documents ICSRs are complete information provided by the reporter at a point in time to describe an event or incident of concern. The report may include information relating to a subject or a group of subjects.
According to Chinese Guidelines for the Collection and Reporting of Individual Adverse Drug Reactions1 A valid report is defined to be consisted of following four elements:
- Identifiable patient
- Identifiable reporter
- Suspected drug
- Adverse reaction.
If all four elements are incomplete, the report is considered invalid and should be added before reporting. “Identifiable” means that the presence of the patient and the reporter can be confirmed. A patient is considered identifiable when one or more of the following are available: name or initials, sex, age (or age group, e.g., adolescent, adult, elderly), date of birth, other identifying code for the patient. The initial reporter who provided the case information or the relevant person contacted to obtain the case information should be identifiable. For case reports from the Internet, the identifiability of the reporter depends on the ability to verify the existence of the patient and the reporter, e.g., by providing a valid email address or other contact information.
Reference:
1. Guidelines for the Collection and Reporting of Individual Adverse Drug Reactions, Announcement by the NMPA on the Issuance of Guidelines for the Collection and Reporting of Individual Adverse Drug Reactions (No. [2018] 131)
Sources & Collection of Adverse Events in China
According to China GVP1, cases are collected from different sources:
- Medical Institutions – Approach like visits, email, telephone, and fax to collect information on AE occurring in clinical settings from medical personnel on a regular basis, keep detailed records, and establish and maintain an adverse drug reaction information file. Doctors could also inform the AE information collected from the patients to the Medical representative who subsequently inform the MAH.
- Drug Distributors – The holder or its distributor should ensure that the pharmaceutical retailer is aware of the effective ways to report AE to it, develop an AE collection plan, and train the pharmacist or other personnel based in the pharmacy, I.e., Pharmacist received AE information feedbacked from the patient that purchased drug on this pharmacy. The pharmacy then send email to PV public mail or call the hotline number for AE complaint.
- Phone calls & Complaints – Calls from the patient that saw the hotline indicated in either Package Insert or outer packaging of the drug or on the MAH websites.
- Academic Literature – AE information identified through literature filtered in literature databases (from literature screening process)
- Internet Relevant Channels – MAH should regularly visit the websites they sponsor or manage to collect possible cases of AE. In principle, MAH are not required to search external websites, but if MAH are informed of AE in external websites, they should assess whether to report them. The MAH should use the company website/portal to collect information on AE, i.e., by creating a dedicated pathway for ADR reporting on the website, providing guidance on how to report, reporting forms and reporting content, and publishing complete and up-to-date product instructions. Graphic media, digital media, social media/platforms initiated or managed by the MAH are also a source of individual adverse drug reactions, i.e., collected using corporate social medial (i.e. Instagram or WeChat public accounts, microblogs, forums, etc.
- Post-marketing Research & Projects – Individual AE identified in post-marketing studies (including those conducted outside of China) or organised data collection projects initiated by the company should be reported as required, such as clinical trials, non-interventional epidemiological studies, focused drug monitoring, patient support projects, market research or other marketing projects, etc. AE identified in post-marketing studies or projects should, in principle, be reported by the MAH to the regulatory authority, but the holder shall not interfere with the reporting behaviour of the study or project collaborators for any reason or by any means.
- Regulatory Authority – If consumer or healthcare professional directly report adverse events to regulatory authority (i.e., FDA, EMA etc..) not to marketing holder (pharmaceutical company), then those reports will be shared to marketing holders by regulatory authorities. This type of cases which were received by companies from regulatory authority are to be handled as regulatory authority cases. Feedback cases downloaded from the ADR monitoring system.
Reference:
1. China Good Pharmacovigilance Practice Announcement of the NMPA on the Publication of China Good Pharmacovigilance Practice (No. 65 of 2021)
Spontaneous AE Reporting in China
A spontaneous report is a direct communication of adverse event information by healthcare professionals or consumers to a company, regulatory authority, or other organization (e.g., WHO, Regional Centres, Poison Control Centre).
Reporting to Pharmaceutical companies: We can even report events directly to manufacturer of product by sending emails/fax or by telephone (details are available in google), some companies have call centres to collect adverse events.
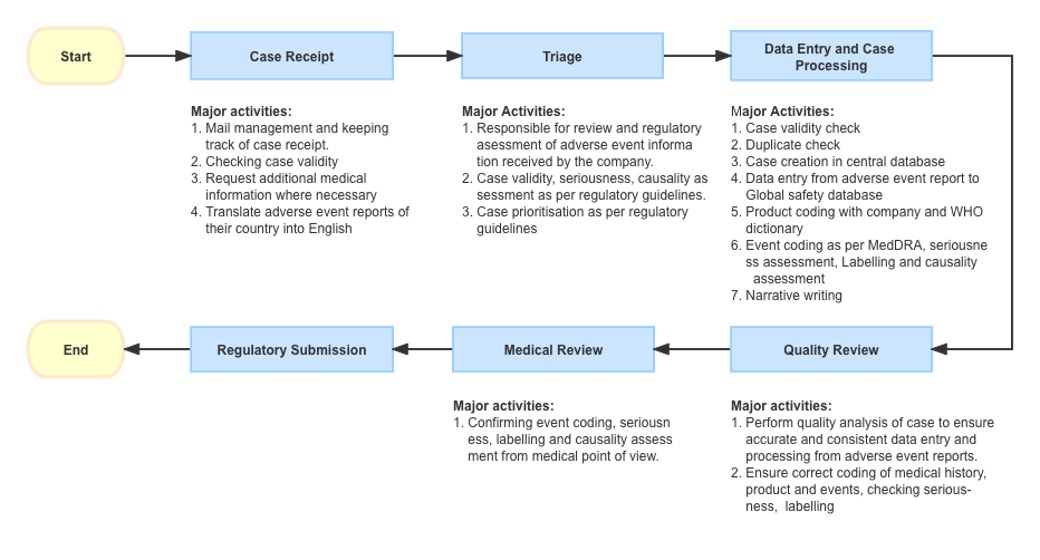
The Call centre personnel received reporting of an AE/AEs from call or an email from a patient’s relative. After an initial confirmation of the case authenticity and validity assessment i.e., the report has identifiable reporter information, an identifiable patient, suspect product/s and AEs, the report will be triaged to PV personnel for further processing.
PV team collect the reported events and perform below assessments (using global sponsor’s database for processing and manual submission to RA):
- Initial seriousness of the event was evaluated
- Assess the reason for event occurrence
- Understanding if it is new event or already known information
- The source document that includes all the communication contents and reporting information will be translated (if necessary)
- Input collected report information in the PV safety database and attaching the source doc + exercise due diligence schedule to collect missing if necessary
- Fill out the information collected as shown below (ArisG System database used for example)
- General information, patient information, protocol/project information (for Research and clinical trial products), product information, adverse event, reporter information
- After initial review of the case processing workflow, the report will be going to next workflow where it is further reviewed and narrative will be written in medical format, the events are coded using Medical Dictionary for Regulatory Activities (MedDRA), causality is assessed, eventually releasing completed CIOMS I form.
- Submitting information to regulatory authority based on the translated CIOMS form
- Save the receipt of submission report (pdf) in local file for inspection/audit purposes.
- Update the tracker.
Feedback Case Reporting in China
Log in the ADR monitoring system.
Click the feedback data button and download the feedback data in excel form
Translate into English to generate an ICSR with all the safety information (if applicable), Input in the PV Safety database and then following the process the same as that of Spontaneous case reporting until the submission to RA completed.
Regulatory Submission
Per the GVP requirement, Spontaneous and feedback case reports can be submitted through ADR monitoring system either manually or through the National Safety Database that is completed interface with the ADR monitoring system.
Medical Review & Narrative Writing
Medical review and narrative writing are crucial components of pharmacovigilance. Medical review involves analysing the safety data generated from clinical trials or post-marketing surveillance to identify potential safety signals and assess the overall benefit-risk profile of a drug. This process requires a deep understanding of the drug’s mechanism of action, clinical pharmacology, and the medical condition it is intended to treat. Medical reviewers must be able to critically evaluate adverse event reports and determine their causality, seriousness, and expectedness. They also need to assess the impact of the adverse event on the patient’s health and well-being and determine if any additional monitoring or risk minimization measures are necessary.
Narrative writing involves summarizing the relevant clinical and safety information in a concise and coherent manner to provide a clear picture of the patient’s medical history and the circumstances surrounding the adverse event. A well-written narrative should provide a chronological account of the patient’s medical history, including any relevant comorbidities, concomitant medications, and previous drug exposure. It should also describe the onset and progression of the adverse event, including the time to onset, duration, and severity, as well as any interventions or outcomes. The narrative should be clear, concise, and free of ambiguity or unnecessary detail.
Effective medical review and narrative writing are critical for identifying and managing potential safety risks associated with drugs. Medical reviewers and writers must be highly trained professionals with a strong background in pharmacology, clinical medicine, and data analysis. They play a vital role in ensuring the safety of patients and the continued success of drug development programs.
Should you have any queries or seeking for consultation, please contact us via online chat or email.